1. 引言
新药设计通常基于体内重要的蛋白质–蛋白质相互作用位点,其中相当一部分是由柔性多肽所介导的,在活细胞中,它们控制了众多至关重要的生理过程[1] 。目前,蛋白质–多肽相互作用已经在制药、生物技术(如蛋白质功能检测) [2] 、生物标记物、生物传感器[3] 以及多肽药物治疗等领域得到广泛应用[4] 。因此,预测和设计多肽–蛋白质相互作用对于生物技术和新药发现具有重要的意义。高分辨率的结构解析法已经在多肽–蛋白质的晶体结构方面得到了广泛应用,包括:核磁共振法和X射线衍射法[5] 。然而,蛋白质结晶过程(包括蛋白质纯化和条件选择)仍存在很大的困难和挑战,而且实验周期长,花费昂贵。
为解决上述问题,可以通过计算模拟的方法来直接预测蛋白质–多肽相互作用。分子对接是预测蛋白质–多肽相互作用最重要的计算技术之一。目前,相关的对接软件包括:AutoDock[6] ,RosettaDock[7] 和DOCK[8] 等等。但是在完全未知互作信息的前提下,进行蛋白质–多肽的相互作用预测尚没有较为系统的方法。AutoDock是盲对接常用的一种软件,其所适用的多肽全长上限仅仅为四个[9] 。另外一种盲对接方法能够克服上述缺陷,然而在入选例子中,互作位点一般均在最大和次大的口袋中[10] 。另外,在许多例子中,非结合态的蛋白质结构通常没有明显的口袋出现在结合位点附近[11] 。
此外,在计算结构生物学中,预测多肽的结合构象是最具有挑战性的,主要因为,多肽具有很大的自由度[12] ,再加上有很多自然状态下的蛋白质并没有稳定的构象[13] ,其内部构象也可能会迅速地发生转变[14] 。有研究表明,考虑多肽的柔性能够增加对接预测的准确性[15] [16] 。同时,相关研究显示,Rosetta FlexPepDock模块能够边实现多肽的从头预测,边实现对接,但是必须已知部分互作信息[7] 。为了解决上述难题,我们提出了一种新的盲对接方法,即在未知互作信息的前提下,用Rosetta FlexPepDock程序实现相互作用位点的系统预测,其整个研究流程见图1A。
2. 材料与方法
2.1. 多肽–蛋白质复合物结构的准备
用于对接的蛋白质–多肽复合物的结构均来自PDB (Protein Data Bank)蛋白质库(见表1)。对于每一
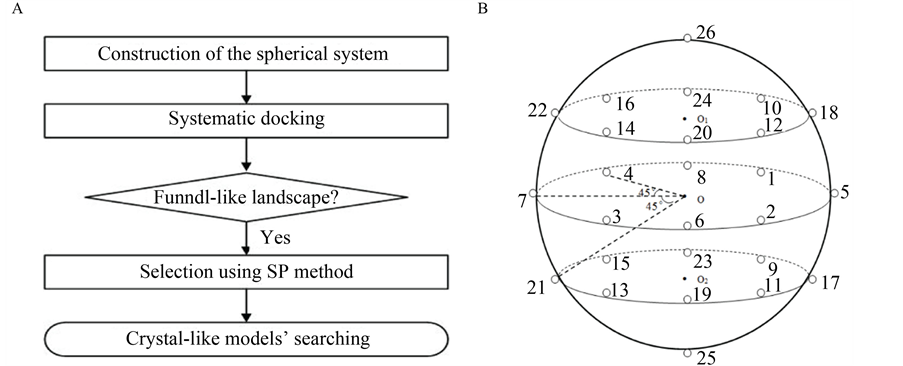
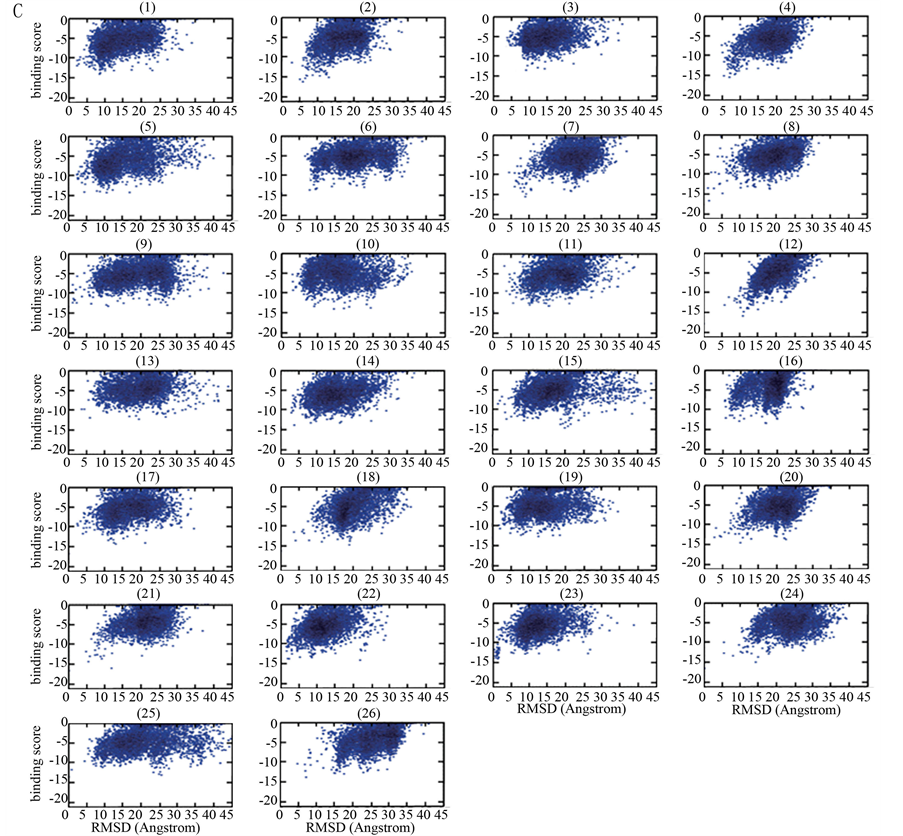
Figure 1. (A) Flowchart of the global docking approach for protein-peptide interactions; (B) The sketch map of the systematic docking with 26 initial locations; (C) The bindingscore-RMSD landscapes with the example of Calmodulin-MLCK docking pair
图1. (A) 多肽–蛋白质相互作用的全局对接流程图;(B) 全局对接系统示意图(26个初始位置);(C) 以Calmodulin-MLCK为例的bindingscore-RMSD散点图

Table 1. Structures of protein-peptide complexes for systematic docking
表1. 系统对接的多肽–蛋白质复合物结构
1表示在实现对接时,将来自3MI9的Tat多肽也考虑进来,因为有关研究显示,Tat多肽和AFF4也有直接的相互作用[18] 。
个结构,首先将其离子、配体以及水分子去掉。另外,如果蛋白质中存在不连续的片段,使用经典建模程序MODELLER将其补齐。从蛋白质–多肽的复合物结构中,选取连续9个氨基酸片段的多肽作为对接多肽片段。原因如下:在Rosetta FlexPepDock中,用于对接的多肽平均长度是9个[7] ,另外,在分析蛋白质序列的疏水性时,其默认的窗口序列长度也是9[17] 。那么,9个连续的多肽片段可以视为性质较为稳定的结构单元。
2.2. 球状对接系统的构建
在未知互作信息的前提下,Rosetta程序通常不能准确预测蛋白质–多肽相互作用位点。为了找到对接的最佳初始位置,通过编写Python脚本来实现全局对接系统的构建:以蛋白质为球心,将多肽均匀地放置在球面26个初始位置上。Rosetta程序中的FlexPepDock模块可以同时实现对接和从头预测,为简化模型,同一多肽在26个初始位置上的构象均设置为相同(图1B)。
2.3. 构象筛选的经验参数法
基于上述构建的系统实现平行对接,并设置相关的参数[19] ,每个位置对接产生5000个构象,并把这些对接构象按其结合自由能(即结合亲和性)大小进行排序:结合自由能越低,排序越前。在这里,结合自由能值直接使用Rosetta经验能量函数值表示,即Rosetta程序计算所得的binding score。按常规的配体–蛋白对接分析过程[7] [20] ,以最低binding score的构象为参考构象,计算得到其它构象对参考构象的RMSD (均方根偏差)值(参考构象的RMSD = 0)。以构象的RMSD值为横坐标,其binding score为纵坐标,获得关于5000个对接构象的binding score-RMSD散点图。
为从26个初始位置中确定出多肽的最佳对接初始位置,我们对binding score-RMSD散点图进行了深入分析,不断摸索与总结,获得了一个筛选最佳初始位置的经验公式。其过程是:对从某一初始位置出发对接所得的5000个构象,把binding score排在前20、且其RMSD值大于0且小于7Å的构象取出,构成一个构象总数为N (0 £ N < 20)的子集, 并设子集中最小RMSD值RMSDmin,对应构象在子集中binding score排序为Rankmin。我们发现,这些从散点图中获得的特征参数存在以下规律:N、RMSDmin和Rankmin越小,binding score也越小,越有利于筛选出好的初始对接位置。因此,有如下用于筛选最佳初始位置的经验公式:
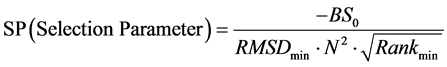 (1)
(1)
上式中的BS0是5000个构象中的最低binding score值,因为5000个对接构象中绝大部分的binding score为负值,所以这个最小值是小于零的,为负值。还有,我们发现上式中的N 对于最佳位置的筛选作用较大,为强化它的作用,将其做平方处理;类似地,Rankmin对于最终结果作用较小,将其进行开方处理。
这样,利用每一个对接初始位置所获得的5000个构象,如果其N > 0,可用上式确定计算该位置对应的SP值,最大SP值的位置即为最佳对接初始位置。对N = 0的位置,不需计算而直接认定其为非最优位置。另外,在与晶体结构对比中,对接能量排在前10低的构象当中,其Cα-RMSD在5.5 Å以内的构象被视为成功预测构象。
2.4. 应用于小分子的ISR法
ISR的全称即固有专一率,ISR法是一种适用于小分子–蛋白质全局对接的定量虚拟筛选法[21] ,它表示对接双方的特异性高低。这种ISR方法适用于小分子化合物与蛋白质的对接[22] 。计算公式如下[21] :
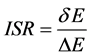 (2)
(2)
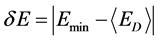 (3)
(3)
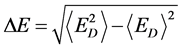 (4)
(4)
δE指的是能隙,即最小能量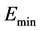 与构象的平均能量
与构象的平均能量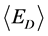 的差值,
的差值,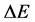 指的是构象的能量波动。
指的是构象的能量波动。
ISR法通过ISR-Affinity散点图来进行直观展示,将本研究提出的SP法与ISR法进行比较,进一步证明其可靠性。
3. 结果与讨论
3.1. Bindingscore-RMSD散点图的经验参数筛选
为了找出正确的或者近似的初始位置,基于收敛状散点图(以Calmodulin-MLCK为例,见图1C),按照经验公式计算筛选参数。根据经验公式(1),将符合公式适用条件的初始位置筛选出来,具体地包括:位置2,4,8,10,12,18,25 (PKA-PKI),位置7,9,10,12 (CycT1_Tat-AFF4),位置2,5,8,11,13,17,18,23 (LAS17-SLA1),位置4,5,7,10,14,15,19 (Androgen-FxxLF),位置2,6,12,16,17,18,19 (Chymotrypsin A-TATI),位置1,2,3,4,5,8,11,12,15,16,18,23,24,26 (ABL1-7C12),位置1,2,3,4,8,12,21,24 (Calmodulin-MLCK),位置2,5,10,12,18,24,26 (SLAM-CD150)。对于上述每个对接例子的粗略筛选位置,计算相应的SP值,最大SP值所在的初始位置即最佳初始位置,见表2。结果显示,PKA-PKI的最佳初始位置是10 (SP value =1.646),CycT1_Tat-AFF4的最佳初始位置是7 (SP value = 0.800),LAS17-SLA1的最佳初始位置是2 (SP value = 1.487),Androgen-FxxLF的最佳初始位置是15 (SP value = 1.543),Chymotrypsin A-TATI的最佳初始位置是19(SP value = 1.751),ABL1-7C12的最佳初始位置是23 (SP value = 1.021),Calmodulin-MLCK的最佳初始位置是24 (SP value = 1.142),SLAM-CD150的最佳初始位置是26 (SP value = 1.376)。
3.2. 基于初始位置的构象选择
基于筛选得到的初始位置,计算结合自由能排在前10低的构象的Cα-RMSD值(与晶体结构相比,见表3)。与晶体结构相比,其中6个例子的最终Cα-RMSD值在5.5Å以内,被视为正确的预测构象,包括:Chymotrypsin A-TATI,Androgen-FxxLF,LAS17-SLA1,Calmodulin-MLCK,ABL1-7C12和CycT1_TatAFF4。只有PKA-PKI和SLAM-CD150 (Cα-RMSD分别为7.71 Å和8.05 Å)不在正确预测的范畴内。总之,
Table 2. The SP value results docking from 26 initial locations of each protein-peptide pair
表2. 每对蛋白质–多肽基于26个初始位置的SP值
Table 3. The lowest Cα-RMSD with respect to the crystal structure, binding score and rank of the binding score
表3. 与晶体结构相比的最小Cα-RMSD值,binding score及其排序
大多数例子的预测结果是成功的,见图2A。
统计数据显示(见图2B),在结合自由能TOP1的构象中,预测正确率为25%,包括CycT1·Tat-AFF4和Calmodulin-MLCK,对应的Cα-RMSD值分别是2.10 Å (Rank1)和3.55 Å (Rank1)。在结合自由能TOP2的构象中,预测正确率为50%,包括CycT1·Tat-AFF4,Androgen-FxxLF,Chymotrypsin A-TATI,Calmodulin-MLCK,对应的Cα-RMSD值分别是2.10 Å (Rank1),4.03 Å (Rank2),5.24.Å (Rank2)和3.55 Å (Rank1)。在结合自由能TOP5的构象中,预测正确率为50%,包括CycT1·Tat-AFF4,Androgen-FxxLF,Chymotrypsin A-TATI,Calmodulin-MLCK,对应的Cα-RMSD值分别是2.10 Å (Rank1),4.03 Å(Rank2),5.24.Å (Rank2)和3.55 Å (Rank1)。在结合自由能TOP10的构象中,预测正确率在75%,包括CycT1·Tat-AFF4,LAS17-SLA1,Androgen-FxxLF,Chymotrypsin A-TATI,ABL1-7C12,Calmodulin-MLCK,对应的Cα-RMSD值分别是2.10 Å (Rank1),5.35 Å (Rank9),4.03 Å (Rank2),5.24 Å (Rank2),5.16 Å (Rank10)和3.55 Å (Rank1)。因此,SP法能够成功识别天然结合构象。当然,为了能够更加全面地检验该方法的可靠性,需要更多的例子加以验证。
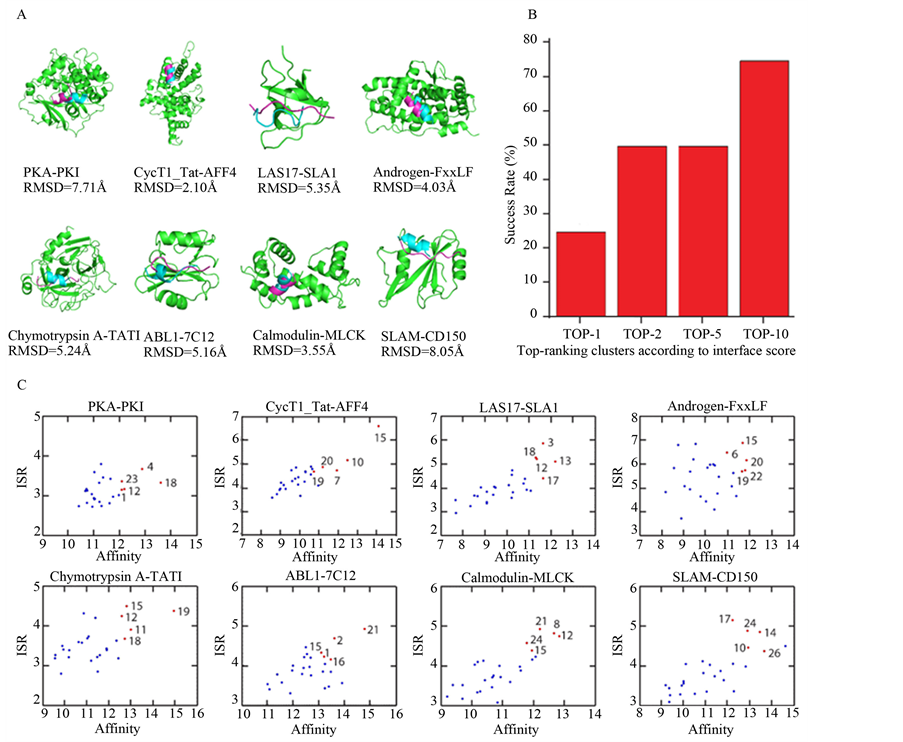
Figure 2. (A) The crystal and predicted conformations. Shown are the native binding pose (magenta) and the final predicted binding pose (blue); (B) The percentages of near-native models of Cα-RMSD values below 5.5 Å with respected to crystal structure in binding score poses of top 1, top 2, top 5 and top 10; (C) ISR-Affinity plots of the eight docking pairs
图 2. (A) 晶体构象(紫色)和预测构象(蓝色);(B) 结合自由能的排序分别在top 1, top 2, top 5 和top 10,近似天然结合构象(与晶体结构相比,Cα-RMSD在5.5 Å以内的构象)的预测成功率;(C) 八个例子的ISR-Affinity散点图
Table 4. Comparison of SP selection results those of ISR selection
表4. SP法与ISR法比较
注:粗体划线的位置表示SP法与ISR法的精准匹配,斜粗体位置表示SP法与ISR法的模糊匹配。
3.3. 与ISR法的比较
最终结果显示,SP筛选法能够在大多数例子中找到最优构象。其他相似的研究揭示,ISR值能够反映小分子–蛋白质对接的特异性。通过对比SP筛选法与ISR筛选法,我们找到了两者间存在的合理关系。在ISR-Affinity的散点图中,处于右上方的点,其ISR值以及亲和性都是最大的。在8个例子当中,SP法选出的点分别在ISR-Affinity散点图的右上方能够找到准确或者模糊的对应(图2C,表4)。与ISR法相比,准确预测结果包括:CycT1_Tat-AFF4(位置7),Androgen-FxxLF(位置15),Chymotrypsin A-TATI(位置19),Calmodulin-MLCK(位置24),SLAM-CD150(位置26)。模糊预测结果包括:PKA-PKI(位置10与ISR法预测的位置1相邻),LAS17-SLA1 (位置2与ISR法预测的位置12相邻),ABL1-7C12 (位置23与ISR法预测的位置15相邻)。结果显示,SP法与ISR法具有62.5%的精准匹配率和100%的模糊匹配率。此外,与SP筛选法相比,ISR筛选法得到的结果并不精准,它能够在前5个结果中找到较为匹配的位置,无法直接找到最优的位置。因此,SP筛选法具有更大的精准度和优势。
4. 结论
本研究提出了一种多肽–蛋白质系统盲对接法,将多肽均匀地分布到以蛋白质为中心的球面上实现平行对接,并提出了经验参数筛选法(即SP法)来筛选预测构象,最终结果显示,多数多肽的预测构象的Cα-RMSD在5.5 Å以下,得以成功识别和预测多肽–蛋白质的相互作用,得到成功的预测结合构象。另外已被证明,我们所提出的这种新的研究方法,与适用于小分子–蛋白质对接的ISR法相比,具有高度的一致性,而且可以将预测结果的范围更进一步地缩小。综上,在未知任何互作信息的前提下,利用该方法能够成功预测蛋白质–多肽的结合位点,在蛋白质与多肽结合位点的预测方面,所发展的方法将有很好的应用价值。
项目基金
上海市重点学科建设项目(B111)。