1. 引言
卟啉和金属卟啉配合物在医学[1] 、分析化学[2] 、光电材料[3] 、人工开发太阳能[4] 及仿生催化氧化[5] [6] 等领域中有非常广泛的应用,有关卟啉化合物合成方法的研究一直以来是国内外的研究热点。其中,四苯基卟啉合成方法的研究较为活跃,本文总结了国内外有关四苯基卟啉合成方法和反应机理的研究进展。
2. 卟啉的结构和性质
2.1. 卟啉化合物的结构
卟吩(图1)是由亚甲基将4个吡咯环连接在一起的高度共轭的平面大分子,具有芳香性,在400 nm附近有较强的吸收带。连接四个吡咯的碳(5,10,15,20位)称为中位(meso)碳,四个吡咯环上共有8个可被取代的碳,称作外环碳。卟吩环上的氢原子被取代后的化合物统称卟啉(porphyrin),根据取代基团的位置不同将卟啉化合物分为两类:一类是在吡咯环上的氢取代,称为α-取代卟啉;另一类是在中位碳上的氢被取代,称为β-取代卟啉。当β位的氢被苯环取代后为四苯基卟啉。
2.2. 卟啉化合物的性质
2.2.1. 物理性质
卟啉是一类颜色较深的固体化合物,具有较高熔点(一般大于300℃),多数卟啉溶液有荧光性且热稳定性好。除此之外卟啉化合物还具有芳香性高,稳定性好,光谱响应宽的特点[7] 。卟啉按溶解性可分为水溶性卟啉和非水溶性卟啉。水溶性卟啉如磺酸基卟啉、氨基卟啉、羧基卟啉等,通常溶于水、甲醇、
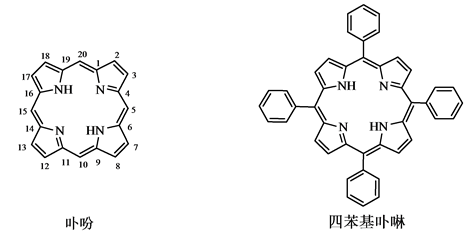
Figure 1. Structure for porphine and tetraphenylporphyrin
图1. 卟吩和四苯基卟啉的结构
丙酮、乙腈等亲水性有机溶剂中,而非水溶性卟啉如四苯基卟啉、对氯四苯基卟啉等一般能溶解于苯、二氯甲烷、吡啶、二甲基甲酰胺等溶剂。
2.2.2. 化学性质
卟啉最显著的化学特性就是易与多数金属离子能生成1:1的配合物。由于卟啉环的中心是一个大空腔,空腔中心到四个氮原子的距离为204 pm,这一数值与第一过渡态金属原子和氮原子的共价半径之和恰好相匹配,因此卟啉化合物极易与过渡金属离子形成稳定的金属配合物,环中心的氮原子上的孤对电子比较容易与金属离子配位[8] 。同时由于卟啉环内的N-H质子具有酸的特性,可以去质子化而变成阴性离子,具有这种平面π共轭电子结构以及大小比较稳定的中心空腔结构的二价离子,非常容易与金属离子结合。
3. 四苯基卟啉的合成方法
卟啉的合成方法主要分为两大类:(1) 非卟啉前体合成卟啉;(2) 对已经形成的卟啉母核进行官能团的修饰合成卟啉。非卟啉前体合成卟啉的方法按照缩合成环方式的不同大致可以分为四吡咯单体直接缩合法和模块法。直接缩合法主要适用于合成结构比较简单的对称性卟啉化合物,如四苯基卟啉和取代四苯基卟啉;模块法则适用于合成些结构复杂、卟啉环上有众多取代基的卟啉化合物。本文对四吡咯单体直接缩合合成卟啉的方法进行概述。
3.1. 经典合成方法
1936年Rothemund[9] 最早提出卟啉(TPP)的人工合成方法:以吡啶为溶剂,以甲醛、乙醛、苯甲醛等醛类化合物和吡咯为原料,将等摩尔的醛和吡咯置于密封容器中,在150℃以下加热反应24~48小时,得到的四苯基卟啉。但是存在反应时间长、产率低、反应条件苛刻、能合成的卟啉种类非常有限等较多缺点。Calvin和他的合作者[10] 在研究Rothemund法时,在反应中加入醋酸锌,将卟啉的收率从4%~5%提高到10%~11%。
1967年,Adler[11] 改进Rothemund的合成方法:在敞开的环境中,以丙酸为溶剂和催化剂,苯甲醛和新蒸的吡咯回流反应30 min,得到四苯基卟啉固体。Adler用此方法,合成大约了70种卟啉化合物,产率在20% ± 3%。Adler法相比于Rothemund法,步骤简单,产率高,是一种比较成熟的合成路线,也是目前应用最广泛的一种卟啉合成方法。但是该法存在反应过程中容易产生大量黑状粘稠物,产物中混有3%的二氢卟啉(TPC)等问题。
1987年Lindsey[12] 提出分步合成卟啉的方法:在氮气保护情况下,以二氯甲烷为溶剂、BF3-乙醚为催化剂,在室温下反应生成四苯基卟啉原,再用1,2-二氯-5,6-二氰对苯醌(DDQ)将四苯基卟啉原氧化为四苯基卟啉,收率可达30%~40%。此方法相比于Adler法,最大的特点在于反应温和,不会产生黑状粘稠物,产物分离容易,可用于合成含有敏感基团的卟啉。但是反应只能在低浓度下进行,以吡咯计仅为0.01 mol/L,且最大反应容积为1 L,反应规模放大后效果不佳。1994年,Lindsey等[13] 研究了在高浓度下的反应(吡咯计,大于0.1 mol/L),采用一步法将原料、氧化剂、催化剂同时加入,简化实验操作步骤,但是最终收率降低,只有10%~20%;
1991年郭灿城等[14] [15] 报道了以无水AlCl3为催化剂,在溶剂DMF中回流反应2 h的合成四苯基卟啉新方法,收率在30%左右。该方法不需要需氮气保护,产物中不含副产物二氢卟啉(TPC),适合于合成含有敏感取代基的卟啉。但是AlCl3易与水反应,生产Al(OH)3沉淀,给产物的分离造成困难。
3.2. 反应溶剂和催化剂的改进
3.2.1. 单一溶剂体系
Rothemund合成卟啉[9] 时以吡啶为溶剂,但是产率通常只有4%~5%,如何提高卟啉产率是此后的研究重点。1964年Adler[16] 提出了可能的反应机理,直到1967[11] 年他提出的以丙酸为溶剂和催化剂的合成方法才解决了卟啉的合成问题,但是也只有20%左右的产率。郭灿城等[14] 以DMF为溶剂,无水AlCl3为催化剂的合成方法将产率提高到了30%。
何明威[17] 改进Lindsey法,以乙醚为溶剂、取代乙酸为催化剂,先在室温下反应2 h,再加入氧化剂硝基苯,在回流温度下继续反应2~3 h,最终产率为40%~60%。该方法产率高,不需要惰性气体保护,不用脱水剂。
高德等[18] 曾用改进的Lindesey法合成了meso位含有强推电子基团的四-(4-(N,N-二甲基)苯胺基)卟啉,他以氯仿为溶剂,三氟乙酸为催化剂,反应1 h以后,加入氧化剂四氯苯醌,继续反应1 h,最终的产率为23%。同时改进了水溶性四-(4-(N,N,N-二甲基-丙磺酸基)苯胺基)卟啉:以DMF为溶剂,回流反应3 h,产率为80%。
1994年潘继刚等[19] 以氯苯、二甲苯、苯甲醚、硝基苯等为溶剂,用间硝基苯甲酸、乳酸、氯乙酸等强度合适的酸作催化剂,吡咯和(取代)苯甲醛反应2~3 h,产率为40%~55%。同时,研究了溶剂、催化剂对卟啉合成反应的影响,发现H+在反应过程中起着催化剂的作用。酸性弱的催化剂,催化能力弱,反应速度慢,产率低甚至得不到产物;酸性强的催化剂易使吡咯质子化,生成吡咯的黑色直链聚合物,不利于生成目标产物,用pKa在2.0~4.0的酸作催化剂,合成产率较高。另外溶剂的极性对产率有较大影响,当以氯苯、二甲苯、苯甲醚、硝基苯等为溶剂时产率较高。此后的文献报道[20] -[25] 证明了潘继刚研究的正确性。其中杨彪等[20] 以二甲苯为溶剂,对硝基苯甲酸或水杨酸为催化剂,在回流条件下反应2.5 h,四苯基卟啉产率为55.4%。刘云等[21] 以甲苯或二甲苯为溶剂,对硝基苯甲酸催化,采用溶剂蒸除–添加同步法,四苯基卟啉产率为58.4%。
刘强等[26] 探讨了反应介质、催化剂、反应物浓度及反应时间等几个主要因素对合成四苯基卟啉产率的影响,当在丙酸、DMF和氯苯溶剂中,以无水三氯化铝或三氯乙酸作催化剂,反应物浓度在0.4~0.5 mol/L,反应时间为2.5 h的条件下,可得到15.2%~28%的产率。
刘雁红等人[27] 报道以DMF或DMSO为溶剂,在无水MgSO4的存在下,采用通入价廉易得的CO2代替丙酸作催化剂一步合成了不含卟啉原的卟啉,CO2在逸出液面时,可带走部分生成水,使反应向生成卟啉的方向移动,CO2的通入还可以保护卟啉被氧气深度氧化分解。
3.2.2. 混合溶剂体系
采用常规的单一溶剂合成带有强供电子基团的卟啉时产率会很低,用混合溶剂法可大大提高产率。混合溶剂法即利用两种或两种以上溶剂按一定比例组成混合溶剂的方法合成卟啉化合物。与单一溶剂体系相比,具有优点以下优点:
(1) 根据不同溶剂的配比得到不同pH值范围的混合液,可满足不同结构的卟啉配体合成的不同酸度要求。
(2) 不同溶剂配比可得到不同沸点的混合液,使反应温度易于控制。
(3) 可以加入相比于空气更有效的氧化剂。
乔庆东等[28] 以体积比为5:1的丙酸与异丁酸的混合溶液,在80℃的温度下合成四苯基卟啉,产率为20.6%。
骆开均等[29] 以体积比为7:3的苯与丙酸的混合溶液,微波加热至回流状态反应6 min,合成的meso-四(对羟基苯)卟啉收率为4.8%。以meso-四(对羟基苯)卟啉为原料,在体积比为7:3的苯与丙酸的混合溶液中微波加热8 min,产物5, 10, 15, 20-[对(二甲基-丁氧基)苯甲酰氧基]苯基卟啉的收率为32%。
李忠芳等[30] 研究了反应温度、反应时间和不同配比的丙酸、乙酸、硝基苯的混合溶液对合成meso-5, 10, 15, 20-四(对羟基苯基)卟啉的影响,当以配比(V/V)为丙酸:乙酸:硝基苯 = 2:2:1的混合溶液,在130℃~140℃,反应时间60 min的条件下,产率达到了39.68%。
孙志成[31] 以不同比例的乙酸、丙酸和硝基苯的混合溶液为溶剂,在回流温度下合成了多种对位和邻位取代的四苯基卟啉,收率15.2%~50.1%。
佘远斌等[32] 以吡咯和取代苯甲醛为原料,在由C1~C8两种不同的直链脂肪酸和硝基苯或硝基苯衍生物组成的混合溶剂中回流反应,卟啉产率40.0%~57.8%。
章艳等[33] 在合成meso-四(对羟基苯)卟啉时,以V(丙酸):V(DMSO) = 20:1的混合液为溶剂回流反应2 h,可以将卟啉收率提高至35%。并且发现DMSO的加入可不但能提高卟啉收率,还以抑制黑色粘稠物是生成,提高产品纯度。
郭灿城等[34] 报道了分别以V(丙酸):V(DMF) = 1:1,V(乙酸):V(甲苯) = 1:3,V(丙酸):V(二甲苯) = 3:1,V(丙酸):V(DMF) = 2:3的混合液为溶剂,在回流状态下以氧气为氧化剂,合成了对氯四苯基卟啉,间甲基四苯基卟啉,间甲氧基四苯基卟啉,对硝基四苯基卟啉。各种卟啉的产率在30%以上,通过HPLC分析,纯度在95%以上,副产物二氢卟啉含量降低到2%以下。
王勤波等[35] 以丙酸或者丙酸与苯、甲苯、对二甲苯、环己烷中的一种组成的混合液为溶剂,在回流条件下合成了四苯基卟啉和取代四苯基卟啉,加入的苯、甲苯、对二甲苯、环己烷起到带水剂的作用。卟啉收率26.2%~37.3%,副产物二氢卟啉含量降低到0.9%以下,产品纯度98%以上。
3.3. 氧化剂的改进
研究发现,在合成卟啉的过程中,一般情况下空气中的分子氧是氧化剂。但通常由于氧化不完全,合成的卟啉产物中含有一定量的二氢卟啉,影响产物纯度[16] 。中间产物卟啉原转化为卟啉和二氢卟啉是竞争反应,因此如何选择合适的氧化剂对产物的收率影响很大。Lindsey[12] 以DDQ或TQC为氧化剂,将卟啉原氧化为卟啉。此后研究人员[30] -[32] [36] -[38] 发现以硝基苯或取代硝基苯为氧化剂,合成的卟啉中不含TPC;当溶剂中含有DMSO时,产品中也不含TPC[37] -[39] ,这是因为DMSO达到沸点时,可分解为二甲基硫醚和氧原子,氧原子可将TPC氧化为TPP。
3.4. 加热方式的改进
传统的加热合成卟啉的方法需要在回流的溶剂中长时间反应,副反应较多且产物分离提纯难,这使卟啉化合物的应用受到一定限制。1986年Gedye等[40] 发现微波可显著加快有机合成反应速率,从此微波在合成化学领域迅速得到重视。1992年法国化学家Petit A及其合作者[41] 成功地将微波合成法引入到卟啉的合成中,其方法为:将吡咯和苯甲醛吸附于硅胶上,利用硅胶的酸性催化反应,在微波炉中加热反应10 min后,经过层析柱进行层析提纯,产率为9.5%。
刘云[21] 等探讨了不同催化剂、不同溶剂条件下TPP 的微波合成,发现采用甲苯或二甲苯做溶剂,对硝基苯甲酸做催化剂,微波加热30 min,产率达到42%,反应速度比常规加热提高了2.2倍。胡希明[42] 也研究了不同催化剂、不同溶剂对微波合成TPP的影响,发现微波诱导时间的延长,产物的产率明显增加。骆开均等人[30] 发现在合成卟啉的酯化衍生物时,微波反应条件下延长反应时间不能提高产物收率,反而有大量黑色物质从溶液中析出。赵胜芳[43] 在微波辐射中合成5, 10, 15, 20-四(4-甲氧羰基苯基)卟啉时也发现微波加热时间太长,产物的产率反而降低。陈年友[44] 等报道了以硝基苯为溶剂,氯乙酸为催化剂,以195 W功率的微波间歇加热6 min,产率为36%。刘红涛等[45] 报道了以二甲苯为溶剂、对硝基苯甲酸为催化剂,功率280 W的微波辐射16 min,产率达到了53.6%。宋溪明等[46] 以离子液体为反应介质,杂多酸为催化剂,微波合成四苯基卟啉。克服了有机溶剂挥发性强、毒性大等缺点。
3.5. 规模化生产方法
郭灿城等[34] 报道了一种由吡咯、芳香醛和空气为原料的高产率合成四苯基卟啉和取代四苯基卟啉的方法和设备,包括了合成卟啉的反应、产物结晶分离、溶剂回收部分,由3 m3的聚合氧化反应器、3 m3的结晶分离器和溶剂精馏塔串联组成。该方法和设备能应用于工业规模化生产,卟啉的产率达到30%以上,二氢卟啉含量降低到2%以下,反应溶剂回收率达到95%以上,并且不需要加入其它溶剂。
王勤波等[35] 报道了一种收率高、安全环保、分离提纯简单、产品质量稳定的工业规模化合成卟啉的方法,包括了带有机械搅拌和夹套、釜顶带冷凝回流的5 m3搪瓷聚合反应器,带搅拌和气体分布器的5 m3氧化反应器,过滤洗涤装置,塔釜体积为5 m3、带3节DN 400 ´ 1000塔节的反应溶剂精馏塔和干燥装置。该方法和设备能使卟啉的收率最高达到37%,副产物二氢卟啉含量降低到0.9%以下,产品纯度98%以上。
4. 卟啉合成机理研究进展
1964年Adler[16] 提出在没有金属盐的存在下,反应的总方程式为:
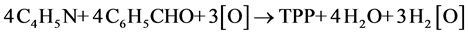 (1)
(1)
反应机理如下:
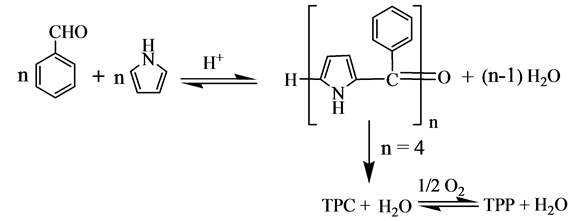 (2)
(2)
在酸的催化作用下,吡咯和苯甲醛生成链长不定的聚合体,当n = 4时生成TPC和TPP,其中n = 4的中间产物生成TPC的步骤是整个反应的速度控制步骤。Adler用此反应机理解释了TPP中含有少量TPC的现象,并指明TPC可以氧化成TPP。但是n = 4时,反应式(2)与总反应方程式(l)的水分子数不相符。如按照反应式(2),在适当的条件下可以得到以TPC为主或者TPC含量高的产物,可是,尚无这方面实验结果的文献报道。因此,上述反应机理欠妥。
1964年,Badger等[47] 提出Rothemund法合成四苯基卟啉的反应机理:
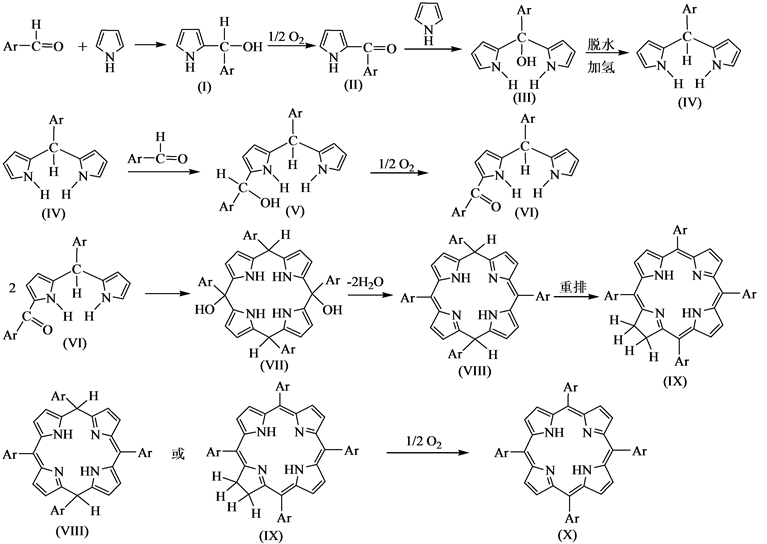 (3)
(3)
Badger提出的反应机理是以带大环化合物(VII)为中间产物,(VII)脱去两分子水变为(VIII),(VIII)经过重排变为二氢卟啉(IX),(VIII)和(IX)可以氧化为最终产物卟啉(X)。该机理能够合理解释生成的卟啉中混有少量二氢卟啉的实验现象,但在适当的条件下应该可以得到 (VIII)和(IX)含量高的产物,可是尚无这方面实验结果的文献报道,并且反应生成总的水分子数和Adler提出的总反应方程式得到的总水分子数不相等。因此,上述反应机理欠妥。
1968年,Adler[48] 深入研究了吡咯和苯甲醛反应生成四苯基卟啉的反应机理,采用分步反应的形式介绍了整个反应历程,其主反应如下:
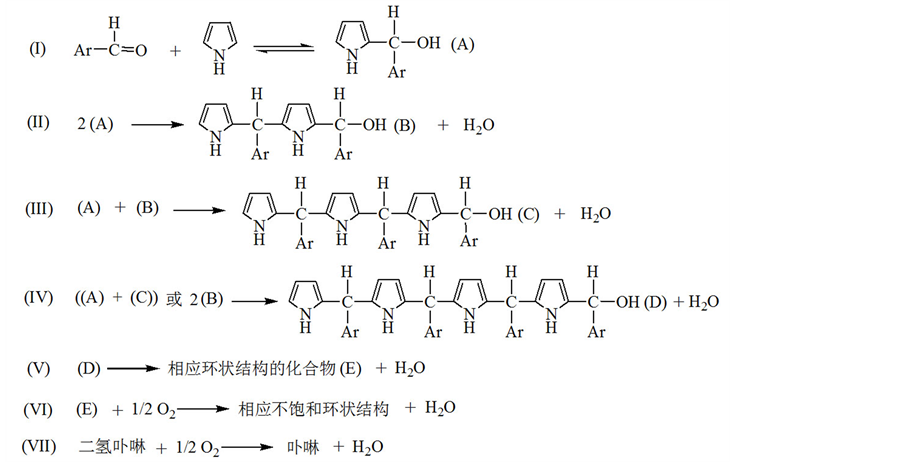 (4)
(4)
在酸的催化作用下,苯甲醛和吡咯生成单吡咯伯醇(I),进而链增长生成四吡咯伯醇,四吡咯伯醇可以环化生成卟啉原,也可以继续链增长生成聚吡咯伯醇,环化过程和生成聚吡咯伯醇的过程是相互竞争的,因此单纯只增大反应物浓度并不能增大卟啉产率,反而会增加长链聚吡咯副产物的量。Adler发现在紫外光谱下苯甲醛的吸收峰迅速消失,表明第一步反应很快,同时水作为反应产物会迅速产生。在有氧气的存在下,卟啉原和二氢卟啉可以氧化为卟啉,同时多吡咯伯醇也会氧化转化为相应的不饱和物质[49] [50] 。但是并没有很好说明二氢卟啉的产生过程,方程式(4)与总反应方程式(l)产生的水分子数也不相等。
1970年Dolphin[51] 利用3,4-二甲基吡咯和苯甲醛的反应相比于吡咯和苯甲醛的反应,在最初聚合阶段速度快,后面氧化阶段速度慢的特征,分离出了卟啉原,证明了卟啉原作为反应重要中间产物的正确性。1971年Kim等[52] 根据实验结果,提出卟啉合成过程中经历的四个重要步骤:
(a) 苯甲醛和吡咯生成单吡咯伯醇
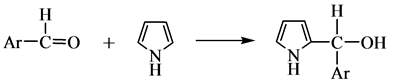
(b) 链增长过程:单吡咯伯醇和另外的吡咯分子、苯甲醛分子反应
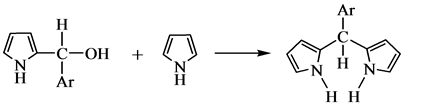
(c) 直链四吡咯化合物的环化生成卟啉原
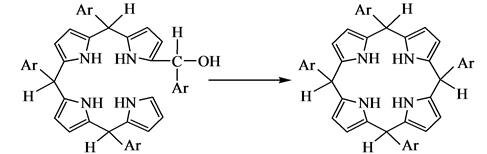
(d) 卟啉原氧化为卟啉

王勤波等[35] 提出的反应机理也证明了卟啉原作为反应重要中间产物的正确性。
1987年Lindsey[12] 认为在有N2的保护情况下,吡咯和苯甲醛在酸的催化作用下生成卟啉原,这一步是可逆的,也是整个反应的速度控制步骤,当加入DDQ等氧化剂时,可将卟啉原氧化为卟啉。根据实验结果,提出的反应机理如下:
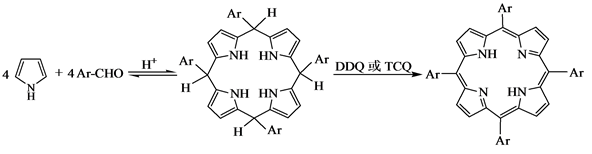 (5)
(5)
郭灿城等[14] 提出的在DMF溶剂中,AlCl3为催化剂合成卟啉的机理如下:
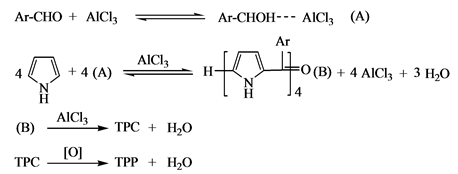 (6)
(6)
郭灿城提出的反应机理与Adler提出的机理基本相同,区别只是AlCl3替代了H+的作用,但是同样存在Adler提出的机理中的问题。
何明威等[37] 参照苯胺和丙烯醛在硝基苯中合成喹啉的Skraup机理,提出了卟啉合成的机理:
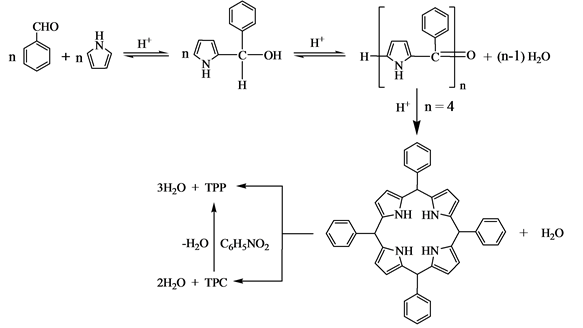 (7)
(7)
反应式的前部分与Adler提出的机理类似,当n = 4时形成卟啉原中间体,卟啉原再转化为TPP和TPC,生成TPP和TPC的反应是相互竞争的,在硝基苯为氧化剂的情况下TPC能转化为TPP。若无硝基苯等氧化剂,合成的TPP中就必然含有TPC。此反应机理可以对实验事实作出合理的解释。
5. 结论及展望
综上所述,目前报道的四苯基卟啉合成方法各有缺点,或反应时间长、产率低;或反应温度高、后处理繁杂;或反应浓度太稀,溶剂用量大。通过对各种方法优缺点对比及成本、合成产率、后处理难易程度和合成规模等多方面考虑,得出合成四苯基卟啉的最佳方法应是Adler法。该法在卟啉的合成过程中,丙酸作为反应溶剂,同时起到催化剂的作用,避免加入其它催化剂,在保证产物收率的基础上,降低了成本,简化了工艺过程,是目前应用最广的四苯基卟啉合成方法。虽然有关四苯基卟啉合成的研究报道很多,但在所用的大多数是挥发性强的有机试剂,绿色合成四苯基卟啉类化合物的文献却很少,如何最大程度减少对环境的危害,找到一条环境友好地合成四苯基卟啉化合物的方法,以满足绿色化学的要求,是今后研究的重点新方向。
基金项目
国家自然科学基金项目(21302049)资助。

NOTES
*通讯作者。