1. 引言
苯并噁唑是一类非常重要的杂环化合物,在医药、农药上有广泛的应用,大量研究表明该类化合物具有广泛的生物活性和药理特征,如有抗菌、抗病毒、抗肿瘤及杀虫的作用 [1] [2] [3] [4] 。因此,对于该化合物的合成及生物活性研究引起了广大药物化学家的极大兴趣。目前已经合成了多种结构的苯并噁唑衍生物,并测试了它们的多种生物活性 [5] [6] [7] [8] 。为了筛选新型具有高效生物活性的药物,合成不同基团修饰的苯并噁唑衍生物仍然是人们热衷的研究课题。
1,2,3-三氮唑是一类很重要的五元氮杂环化合物,广泛用于制药、染料、光稳定剂及防腐蚀剂方面 [9] ,特别是它具有广泛的生物活性,如抗菌 [10] 、抗炎、抗病毒 [11] 、抗肿瘤 [12] [13] 的作用,因而几十年来受到了药物化学工作者的极大关注。近年来,虽然有许多关于1,2,3-三氮唑化合物的制备及其生物活性研究的报道 [14] - [19] ,但是对其中含有1,2,3-三氮唑官能团的苯并噁唑衍生物的合成及抗肿瘤活性研究则罕见报道。鉴于不同活性的基团在同一分子中聚集能明显改善化合物的生物活性这一特性,本文应用活性亚结构拼接原理,将2-巯基-5-甲基苯并噁唑引入到1,2,3-三氮唑化合物中,设计合成系列新的2-(1-取代-1H-[1,2,3]三唑-4-甲硫基)-5-甲基苯并噁唑类化合物,以期从中筛选得到具有显著抗肿瘤活性和应用前景的化合物。而这方面的研究还未见文献报道。合成路线见图1。
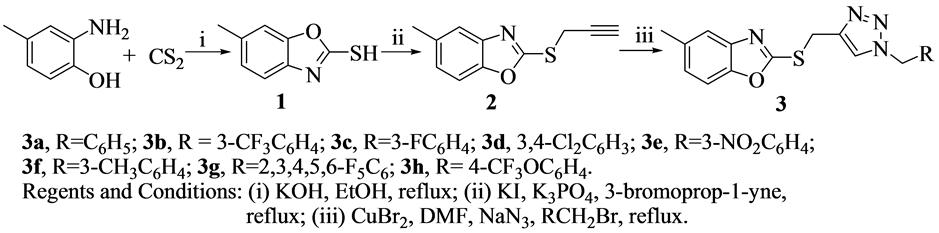
Scheme 1. Synthetic route of compounds 3(a~h)
图1. 化合物3(a~h)的合成路线
2. 实验方法
2.1. 主要试剂与仪器
XRC1显微熔点仪测定(温度未校正);Varian400MHz核磁共振仪(Me4Si为内标,DMSO-d6为溶剂);Finnigan-MAT4510型质谱仪,离子源为ESI;FT-IR169型红外光谱仪(固体用KBr压片,液体用液膜法);Carlo-Erba-1106 型元素分析仪。
其它所用试剂均为市售化学纯或分析纯,除特别注明外,未经进一步处理。
2.2. 中间体1的合成
在50 mL的圆底烧瓶中加入2-氨基-4-甲基苯酚1.09 g (0.01 mol)和二硫化碳3.04 g (0.04 mol),在室温下加入乙醇溶液20 mL和氢氧化钾0.56 g (0.01 mol),加热回流4 h,薄层色谱(TLC)跟踪反应,反应完毕,冷却至室温,加水溶解后,用稀盐酸调PH至6~7,后加入乙酸乙酯萃取,静止分出乙酸乙酯层,有机层用水洗(100 mL × 2),合并有机相,无水硫酸钠干燥,再通过旋转蒸发仪除去乙酸乙酯,得黄色固体粗产品中间体1。用甲醇溶解并以1:1加入硅胶,旋蒸除去溶剂,柱层析,展开剂(V石油醚:V乙酸乙酯 = 3:1),收集溶液,蒸去溶剂,得淡黄色固体,收率92%。1H NMR (DMSO, 400 MHz, ppm), δ: 13.82 (s,1H, S-H), 7.39 (d, J = 8 Hz, 1H, Ar-H), 7.06 (s,H, Ar-H), 7.06 (s, 1H, Ar-H), 2.37 (s,3H, ArC-H). MS m/z (%): 165 (M+, 100).
2.3. 中间体2的合成
称取5-甲基-2-巯基苯并噁唑(中间体1) 0.66 g (4 mmol)、碘化钾0.066 g (0.4 mmol)和无水磷酸钾0.85 g (4 mmol)于50 mL圆底烧瓶中,加入10 mL乙腈溶液,在室温、搅拌条件下缓慢加入溴丙炔0.31 mL (4 mmol),在82℃下加热回流5 h,冷却至室温,过滤,旋蒸除去溶剂,得到棕红色粘稠油状粗产品,粗产品经300~400目硅胶柱层析(洗脱剂:V石油醚:V乙酸乙酯 = 3:1),收集含有中间体2的溶液,旋蒸除去溶剂,得到红棕色油状液体,放置一段时间得到红棕色固体。收率为97.44%。1H NMR(CDCl3,400 MHz, ppm), δ: 7.40 (s,1H, Ar-H), 7.32 (d, J = 8 Hz,1H, Ar-H),7.07(d, J = 8 Hz,1H, Ar-H),4.08(d, J = 8 Hz, 2H, SC-H), 2.45(s, 3H, ArC-H), 2.30 (t, J = 4 Hz, 1H, C≡C-H). 13C NMR (CDCl3, 100 MHz, ppm) δ: 162.8, 150.3, 141.9, 134.2, 125.1, 118.7, 109.3, 75.3, 72.4, 21.41, 20.7. IR (KBr) v: 3315, 1620, 1572(C=N), 628 (C-S)cm-1; MS m/z (%): 203 (M+, 100).
2.4. 目标化合物3(a~h)的合成
在20 mL的反应试管中加入叠氮化钠0.715 g (1.1 mmol)、溴化铜0.0112 g (0.05 mmol),密封并通入氩气,真空泵置换三次,将试管内的氧气排尽,然后准确称取0.1902 g (1 mmol)中间体2,用1 mL DMF溶解,使用注射器缓慢的注入反应试管中,再通过注射器将取代苄溴(1 mmol)缓慢注入反应试管中,在70℃反应5 h,反应完毕,冷却至室温,加水20 mL,乙酸乙酯萃取(15 mL × 3),合并乙酸乙酯层,无水硫酸钠干燥,蒸去溶剂,粗产品经柱层析(洗脱剂:V石油醚:V乙酸乙酯 = 3:1)得到相应的目标化合物3(a-h)。
化合物3a: 白色固体,收率92. 5%.1H NMR(CDCl3,400 MHz, ppm), δ: 7.58 (s, 1H, triazole ), 7.33 (s, 1H, Ar-H ), 7.30 (t, J = 4 Hz, 3H, Ar-H ), 7.24 (d, J = 6.4 Hz, 1H, Ar-H), 7.21-7.18 (m, 2H, Ar-H), 7.01 (d, J = 6.8 Hz, 1H, Ar-H), 5.44 (s, 2H, N-CH2), 4.56 (s, 2H, SC-H), 2.41 (s, 3H, Ar-CH3). 13C NMR (CDCl3, 100 MHz, ppm) δ: 164.0, 150.2, 143.8, 141.9, 134.2, 129.1, 128.7, 127.9, 124.9, 122.9, 118.4, 109.3, 54.1, 30.9, 21.4. MS m/z (%): 336 (M+, 100). Anal. calcd for C18H16N4OS: C 64.26, H 4.79, N 16.65; found C 64.27, H 4.76, N 16.63.
化合物3b: 淡黄色固体,收率87.3%.1H NMR(CDCl3,400 MHz, ppm), δ: 7.70(s, 1H, triazole), 7.44 (m, 2H, Ar-H), 7.28 (s, 2H, Ar-H), 7.21 (s, 1H, Ar-H), 7.13 (d, J = 8.4 Hz, 1H, Ar-H), 6.99 (d, J =8.4 Hz, 1H, Ar-H), 5.43 (s, 2H, Ar-CH2), 4.49 (s, 2H, S-CH2), 2.31 (s, 3H, Ar-CH3). 13C NMR (CDCl3, 100 MHz, ppm) δ: 163.8, 150.1, 144.0, 141.8, 135.7, 134.1, 131.2, 129.6, 125.4, 125.3, 124.9, 124.6(q, J = 30 Hz), 124.5, 123.2, 118.3, 109.2, 53.3, 26.7, 21.2. MS m/z (%): 404 (M+, 100). Anal. calcd for C19H15F3N4OS: C 56.43, H 3.74, N 13.85; found C 56.41, H 3.76, N 13.84.
化合物3c: 淡黄色固体,收率77.3%.1H NMR(CDCl3,400 MHz, ppm), δ: 7.62 (s, 1H, triazole), 7.32 (s, 1H, Ar-H), 7.24-7.23 (m, 2H, Ar-H), 7.04-7.00 (m, 2H, Ar-H), 6.96 (d, J = 8 Hz, 1H, Ar-H), 6.91 (d, J = 8 Hz, 1H, Ar-H), 5.44 (s, 2H, Ar-CH2), 4.51 (s, 2H, S-CH2), 2.43 (s, 3H, Ar-CH3). 13C NMR (CDCl3, 100 MHz, ppm) δ: 163.8, 150.2, 148.3, 144.4, 144.8, 136.6, 134.2, 133.8, 130.2, 125.0, 123.6, 123.2, 122.8, 118.4, 109.3, 53.0, 28.9, 21.4. MS m/z (%): 354 (M+, 100). Anal. calcd for C18H15FN4OS: C 61.00, H 4.27, N 15.81; found C 61.03, H 4.26, N 15.84.
化合物3d: 淡黄色固体,收率74.2%.1H NMR(CDCl3,400 MHz, ppm), δ: 7.63 (s, 1H, triazole), 7.34-7.23 (m, 4H, Ar-H), 7.02-6.98 (m, 2H, Ar-H), 5.39 (s, 2H, Ar-CH2), 4.55 (s, 2H, S-CH2), 2.41 (s, 3H, Ar-CH3). 13C NMR (CDCl3, 100 MHz, ppm) δ: 163.8, 150.2, 144.4, 144.3, 141.8, 134.6, 133.2, 133.0, 131.2, 129.3, 125.0, 123.0, 118.6, 118.4, 109.3, 52.8, 26.9, 21.4. MS m/z (%): 404 (M+, 100). Anal. calcd for C18H14Cl2N4OS: C 53.34, H 3.48, N 13.82; found C 53.37, H 3.46, N 13.84.
化合物3e: 淡黄色固体,收率79.2%. 1H NMR(CDCl3,400 MHz, ppm), δ: 8.11-8.05 (m, 2H, Ar-H), 7.72 (s, 1H, triazole), 7.50-7.42 (m, 2H, Ar-H), 7.23-7.20 (m, 1H, Ar-H), 6.99 (d, J = 2 Hz, 1H, Ar-H), 5.55 (s, 2H, Ar-CH2), 4.55 (s, 2H, S-CH2), 2.38 (s, 3H, Ar-CH3). 13C NMR (CDCl3, 100 MHz, ppm) δ: 163.9, 150.3, 148.3, 144.5, 144.2, 136.5, 134.2, 133.8, 130.2, 125.0, 123.6, 123.2, 122.6, 118.4, 109.3, 53.1, 26.9, 21.4. MS m/z (%): 381 (M+, 100). Anal. calcd for C18H15N5O3S: C 56.68, H 3.96, N 18.36; found C 56.70, H 3.98, N 18.34.
化合物3f: 淡黄色固体,收率86.8%.1H NMR(CDCl3,400 MHz, ppm), δ: 7.55 (s, 1H, triazole), 7.31 (s, 1H, Ar-H), 7.23-7.21 (m, 1H, Ar-H), 7.14-7.07 (m, 4H, Ar-H), 7.01-6.98 (m, 1H, Ar-H), 5.37 (s, 2H, Ar-CH2), 4.53 (s, 2H, S-CH2), 2.40 (s, 3H, Ar-CH3), 2.28 (s, 3H, Ar-CH3). 13C NMR (CDCl3, 100 MHz, ppm) δ: 164.0, 150.2, 143.8, 141.9, 138.6, 134.1, 131.4, 129.8, 128.2, 128.0, 124.9, 122.8, 118.4, 109.3, 101.7, 53.9, 26.9, 21.4, 21.1. MS m/z (%): 350 (M+, 100). Anal. calcd for C19H18N4OS: C 65.12, H 5.18, N 15.99; found C 65.10, H 5.16, N 15.96.
化合物3g: 淡黄色固体,收率98.5%.1H NMR(CDCl3,400 MHz, ppm), δ: 7.55 (s, 1H, triazole), 7.32 (s, 1H, Ar-H), 7.23-7.21 (m, 2H, Ar-H), 5.38 (s, 2H, Ar-CH2), 4.54 (s, 2H, S-CH2), 2.42 (s, 3H, Ar-CH3). 13C NMR (CDCl3, 100 MHz, ppm) δ: 163.8, 150.2, 146.6, 144.2, 141.8, 134.3, 134.1, 125.0, 123.2, 122.8, 118.3, 109.3, 108.2, 53.8, 26.6, 21.4. MS m/z (%): 426 (M+, 100). Anal. calcd for C18H11F5N4OS: C 50.71, H 2.60, N 13.14; found C 50.68, H 2.62, N 13.16.
化合物3h: 淡黄色固体,收率86.8%.1H NMR(CDCl3,400 MHz, ppm), δ: 7.61 (s, 1H, triazole), 7.33(s, 1H, Ar-H), 7.25-7.21 (m, 3H, Ar-H), 7.15-7.13 (m, 2H, Ar-H), 7.04-7.02 (m, 1H, Ar-H), 5.46 (s, 2H, Ar-CH2), 4.57 (s, 2H, S-CH2), 2.42 (s, 3H, Ar-CH3), 2.28 (s, 3H, Ar-CH3). 13C NMR (CDCl3, 100 MHz, ppm) δ: 163.9, 150.2, 149.3(t, J = 2Hz), 141.9, 134.2, 133.1, 129.4, 125.0, 124.2, 122.9, 121.6, 121.4, 118.4, 109.3, 53.2, 26.7, 21.4. MS m/z (%): 420 (M+, 100). Anal. calcd for C19H15F3N4O2S: C 54.28, H 3.60, N 13.33; found C 54.31, H 3.63, N 13.36.
3. 结果与讨论
本文合成的8个2-(l-取代-1H-[1,2,3]-三氮唑-4-甲硫基)-5-甲基苯并噁唑化合物3a~3h均为固体,易溶于氯仿、DMF和DMSO, 长时间放置无明显变化,其结构组成均经过MS、1H NMR、13C NMR和元素分析得到确证,合成的产物即为标题化合物。
3.1. 取代苄溴对目标物制备的影响
5-甲基-2-(2-丙炔硫基)苯并噁唑与取代苄溴及NaN3的反应是一类典型的[3+2]环化反应,即Click反应。实验结果表明:取代苄溴的苯环上连有吸电子基团如NO2、卤素(X), 或是供电子基团如CH3时,反应均容易进行,可以74.2%~98.5%的收率得到目标化合物3(b-h) (表1,entries 2-8)。

Table 1. The reaction of compounds 2 with benzyl bromide and NaN3 catalyzed by CuBr2a
表1. CuBr2催化化合物2与苄溴及NaN3的反应a

a1 mmol 2,1 mmol RCH2Br,1.1 mmoL NaN3,1 mL DMF;b分率产率。
Table 2. The inhibition rates for CDC25B of the compounds 3(a-h) in vitroa
表2. 化合物3(a-h)对CDC25B的抑制活性a
aConcentration:20 μg/mL,Inhibition (%) > 50% (化合物浓度:20 μg/mL,抑制率(%) > 50%),有抑制作用;bStandard drug (标准药物)。
3.2. 目标化合物3的谱图分析
化合物3的1H NMR谱图中,在δ 7.72~7.55 ppm左右出现的单(宽)峰为目标化合物中1,2,3-三氮唑环上的C=C-H氢质子的吸收峰,在δ 6.80~7.50 ppm范围内的多重峰为芳环上的质子吸收峰。在δ 5.55~5.38 ppm左右出现的单峰为目标化合物中Ar-CH2N上的氢的位移,Ar-CH2-trizaole中氢的位移在δ 4.51~4.57 ppm。化合物的MS谱图中,所得的目标化合物均给出分子离子峰,与相应的分子式一致。因此,化合物3为目标化合物。
3.3. 目标化合物3的抗肿瘤活性
抗肿瘤活性采用测试化合物对细胞周期分裂蛋白25B (CDC25B)的抑制率进行初步评价。其测试由上海国家新药筛选中心进行。
其方法是:采用荧光底物OMFP(邻甲基荧光磷酸酯),经CDC25B去磷酸化后得到的产物OMF在被485 nm激发光激发后可发射出波长为535 nm的可检测的荧光信号,从而观察酶的活性变化以及化合物对其的抑制情况。实验中CDC25B所采用的阳性参照化合物为Na3VO4,阴性参照物为DMSO。然后通过测试活性与剂量的依赖关系,用计算软件Graphpad Prism对二者进行非线性拟和得到半抑制浓度(IC50)值。测试结果如表2所示,除化合物3(a, d, g, h)外,化合物3(b,c,e,f)在浓度为20 uM时均显示出中等的抗CDC25B活性,其IC50为13.23~14.68 ug/mL。同时可以看出当苯环上连有吸电子基(F, NO2)时,目标化合物都能表现出良好的抗CDC25B活性。
4. 结论
以2-氨基-4-甲基苯酚、二硫化碳和3-溴丙炔为原料,经环化、取代、Click反应合成了8个新的2-(l-取代-1H-[1,2,3]-三氮唑-4-甲硫基)-5-甲基苯并噁唑化合物3(a~h),初步生物活性测试结果表明,在20 uM 浓度下,部分化合物表现出良好的抗CDC25B活性,其抑制率为可达68.18%,IC50可达13.23 ug/mL。由于测试的模型有限,尚需在今后的研究中进一步的扩展模型进行抗肿瘤活性研究,深入探讨其结构与生物活性之间的关系。因此,2-(l-取代-1H-[1,2,3]-三氮唑-4-甲硫基)-5-甲基苯并噁唑类衍生物可以作为抗肿瘤药物的先导分子进行进一步的研究。
基金项目
西南民族大学研究生创新基金(No. CX2016SZ063),四川省科技厅科技支撑计划项目(No.2015NZ0033)。
*通讯作者。