1. 引言
随着原子尺度沉积技术的进步,人们在钙钛矿氧化物异质结中发现了很多新颖的物理现象。其中最具代表性的就是LaAlO3/SrTiO3异质界面处的二维电子气 [1] [2] [3] [4] 。在生长过程中,根据不同沉积序列,会形成两种LaAlO3/SrTiO3界面。一种是TiO2/LaO界面,称为n型界面;第二种是由SrO/AlO2形成的界面,称为p型界面。尽管这两种材料的块材都是典型的带隙绝缘体,在该(001) n型界面上却存在高迁移率的准二维电子气 [1] [5] 。且该准二维电子气只有在LaAlO3厚度达到临界值1.6 nm (4个LaAlO3元胞厚度)时才会出现 [6] [7] 。只是这些现象和报道都局限在钙钛矿氧化物(001)异质结界面。最近,已经有文献报道了SrTiO3和LaAlO3的(110)和(111)界面上的电荷重构 [8] 。文献还称,在同样存在极化不连续的SrTiO3(111)表面上,即使沉积非晶的LaAlO3,照样可以实现电荷转移。基于杂化密度泛函计算,我们已经在LaAlO3/SrMnO3(001)超晶格中发现了电荷转移和高自旋极化的铁磁金属性 [9] 。那么这一体系的(111)超晶格中是否也会出现类似的现象呢?本文基于第一性原理计算,在LaAlO3/SrMnO3沿[111]方向形成的(LaO3)/Mn超晶格界面上也获得了由于电荷重构而导致的半金属性。在此,电荷从LaAlO3侧转移到SrMnO3侧,占据Mn离子空的eg轨道。我们认为与该体系(001)界面上的电荷转移类似 [9] ,该电荷转移是由纯粹的极化不连续导致的,与电子之间的关联没有直接关系。
2. 模型和计算方法
第一性原理模拟界面时用得最多的是对称的超晶格。在这里,沿着[111]方向把一定数目的SrMnO3元胞与一定数目的LaAlO3元胞排列起来,服从周期性边界条件约束。在这个对称的超晶格中,LaAlO3和SrMnO3都是非化学计量比的。如果SrMnO3含一个多余的SrO3原子层,LaAlO3就多余一个Al原子层,此时由于对称性,就形成两个相同的p型界面,我们称为p型超晶格,如图1(a)所示。同理,Mn和LaO3原子层就形成两个等价的n型界面,如图1(b)所示,称为n型超晶格。
沿[111]方向生长的ABO3钙钛矿结构形成六角晶格,(111)平面内的每个B离子与下一层(111)平面内的3个B离子是最近邻,同样与上一层的(111)平面内的三个Mn离子也是最近邻,而与同一层内的其他等价的4个Mn离子则是次近邻,所以考虑到Mn离子的磁性,平面内元胞取
(a为SrMnO3块材的晶格常数)。计算使用VASP软件包,选用GGA方法处理体系中电子与电子之间的交换关联作用,具体交换关联泛函选择的是PBE。选用缀加平面波(PAW)来处理电子-离子之间的相互作用,把Sr原子的
,Mn原子的
,La原子的
,O原子的
当做价电子处理。平面波展开的截断能(Energy Cutoff)取为600 eV,每个原子上的自洽场收敛条件设为
。对Brillouin区的

Figure 1. Illustration of two kinds of LaAlO3/SrMnO3(111) superlattices: (a) p-type, (b) n-type
图1. 两种SrMnO3/LaAlO3(111)超晶格结构示意图。(a) p型;(b) n型
积分计算使用Monkhorst-Pack方案,以
点为中心选择k点网格为
[10] 。考虑到过渡元素Mn上电子的关联效应,我们选取Mn元素的在位能参数
,计算所用的晶格常数取实验值
[9] 。
3. 结果与讨论
对于图1(a)和图1(b)所示的两种超晶格,我们保持总钙钛矿结构单元数不变,逐渐增加SrMnO3成分所占比例,分别用p-M和n-M来标记我们所用的各种超晶格结构,其中p和n是代表超晶格界面类型,M是Mn层数。以p-2和n-2为例,图1(a)和图1(b)分别给出两种超晶格结构示意图。改变M数值,我们就可以详细讨论量子受限机制的影响,以及体系电子性质和磁性质随Mn层厚度的依赖关系。考虑到电荷转移会引起轨道填充的变化,从而可能会引起氧八面体的畸变,我们用
结构作为初始晶格结构进行弛豫。根据Glazer定义,
结构下的氧八面体畸变类型为
,对比图1(a)和图1(b)可以清晰地看出这种畸变。
3.1. p型超晶格中的空穴转移
我们先来讨论p型LaAlO3/SrMnO3(111)超晶格。表1给出对应各p-M型超晶格的总能量和Mn离子磁矩,其中
,即铁磁态能量与G类反铁磁态能量之差。对于p-1而言,因为超晶格中只有一个Mn离子,所以
记为0。从表1可见,所有p型组合下Mn离子之间的基态磁构型仍然保持了SrMnO3块材的G类反铁磁序,也就是Mn离子无论在ab平面内,还是沿c轴方向与其最近邻都是反铁磁耦合。在这些p型超晶格中,体态中空的eg轨道依然没有被占据。图2给出所计算的四种p-M超晶格每个原子面上的总态密度图。图3给出p-2超晶格的能带结构图,虚线代表费米能级所在位置,上半栏代表spin-up,下半栏代表spin-down,且用玫红色标识出Mn离子的轨道分布。图4给出的是p-2超晶格的自旋密度图。
与块材SrMnO3和LaAlO3都不同,p型超晶格是金属性的,费米面穿过价带顶,不过从图2可以得出,空穴基本上平均分布在超晶格中SrO3面和LaO3面的氧离子上。这与LaAlO3/SrMnO3(001) p型超晶格不同 [9] ,这是由于(001)和(111)超晶格界面处的极化不连续性差别造成的。LaAlO3/SrMnO3 (001) p型界面处,LaAlO3中SrMnO3则没有极化电场,所以LaAlO3中的极化电场会使空穴从LaAlO3侧转移到SrMnO3侧,不过到达SrMnO3侧的空穴不会留在界面上,而是平均分布在SrO面上的氧离子上。而LaAlO3/SrMnO3 (111) p型界面则不同,极化电场同时存在于LaAlO3和SrMnO3侧,只是场强大小有所区别,所以空穴基

Table 1. Total Energies and Local Magnetic Moment (μB/Mn) of p-Type Superlatticesfor Various Magnetic Orderings
表1. 各p型(111)超晶格不同磁构型的能量差(meV/Mn)和局域磁矩(μB)

Figure 2. Partial density of states of every atomic planes of four kinds of p-type superlattices. Fermi level is at energy zero (dashed line)
图2. 四种p型(111)超晶格各原子面上总的电子态密度,费米能级用虚线示意。(a) p-1;(b) p-2;(c) p-3;(d) p-4
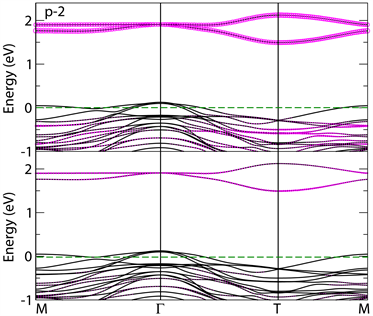
Figure 3. Electronic band structure and Spin density of p-2 superlattice
图3. p-2超晶格的电子能带图
本上是均匀分布于SrO3面和LaO3面的氧离子上。图3的能带图一方面清楚地显示出Mn离子上满占据的t2g和空的eg轨道,另一方面也展示了该体系的金属特性,且金属性来源于氧离子的p轨道。这与图2的能态密度图是相符的。图4是p-2超晶格的自旋密度图,图中两种不同颜色分别代表不同自旋方向,可以清晰看出Mn离子无eg轨道占据特征,只有t2g轨道特征,这进一步验证图2和图3中Mn离子满占据的t2g轨道特征。
3.2. n型超晶格中的电子转移
下面我们来讨论n型(111)超晶格界面处的电子结构。表2给出对应该类型各超晶格不同磁有序结构的总能量之差和对应Mn离子的局域磁矩,其中
,即G类反铁磁态与铁磁态能量之差。对于n-4超晶格我们还计算了其他反铁磁构型,不过能量都相对较高,所以没有在表格中给出。从表2可以看出,虽然SrMnO3块材是G类反铁磁,各n型(111)超晶格界面处均是铁磁序能量最低。图5给出以n-2超晶格为代表的基态(即铁磁态)能带结构图,图中虚线代表费米能级所在位置。图6则为n-2超晶格中每个Mn离子所有价电子的自旋密度图,该图显示了完全自旋极化的t2g态和eg态。与n型(001)超晶格结构类似,所有n型(111)超晶格均显示半金属性,费米面处的巡游电子态主要来自于完全自旋极化的Mn离子。不同的是,(111)超晶格费米面附近出现了比较平坦的能带,我们认为这是六角晶格对称性所决定的,类似的平坦能带也出现在(111)LaNiO3体系中 [11] [12] 。另外,能带图显示n-2超晶格的下自旋
Table 2. Total energies and local magnetic moment ( μ B / M n ) of n-type superlattices for various magnetic orderings
表2. 各n型(111)超晶格不同磁构型的能量差(meV/Mn)和局域磁矩( μ B )
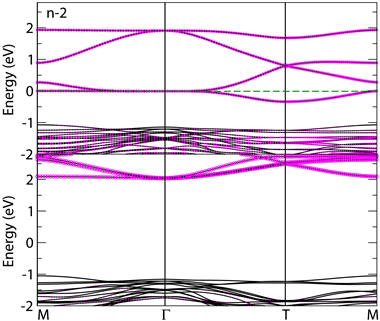
Figure 5. Electronic band structure of n-2 superlattice
图5. n-2超晶格的电子结构能带图

Figure 6. Electronic band structure of p-2 superlattice
图6. n-2超晶格的电子结构能带图和自旋密度图
带带隙为3.0 eV左右,大于(001)超晶格中数值(约2.5 eV),更是远远大于传统掺杂SrMnO3块材的对应值大很多,比如,La0.67Sr0.33MnO3块材对应带隙只有1 eV [13] [14] [15] 。
为了进一步探讨半金属铁磁性的来源,我们在图7中给出所计算的四种n型(111)超晶格各原子面上总的电子态密度,其中虚线对应费米能级所在位置。在SrMnO3块材中,Mn离子是+4价,含3个3d电子,所以t2g轨道是满占据的,eg轨道是空的。而从图7中的电子态密度很明显可以看出,此时所有Mn离子的eg轨道都被部分占据。这些被占据的eg电子态说明在n型(LaO3)3-/Mn4+界面上也出现了电荷转移。从表2中的磁矩数值可以看出,转移过来的电荷并不是均匀分布在每个Mn离子上,这点与n型(001)界面不同 [9] 。另外,从表2还可以看出,随着SrMnO3层数增加,Mn的局域磁矩逐渐减小,但是SrMnO3

Figure 7. Partial density of states of every atomic planes of four kinds of n-type superlattices. Fermi level is at energy zero (dashed line)
图7. 四种n型(111)超晶格各原子面上总的电子态密度,费米能级用虚线示意。(a) n-2;(b) n-3;(c) n-4;(d) n-5
层数固定的超晶格来说,每个Mn离子的磁矩之和基本保持不变,均为1左右,这就说明转移过来的电子数是固定的常数。该n型LaAlO3/SrMnO3(111)超晶格中所发现的二维电子气,与相同体系(001)超晶格中所发现的二维电子气类似 [9] ,都是巡游的eg电子与局域的t2g电子共存,这样也就自然地遵从Zener双交换机制 [16] [17] ,所以出现了半金属铁磁性。
4. 总结
本文利用第一性原理计算预测了p型LaAlO3/SrMnO3(111)超晶格中出现的均匀分布在SrO3面和LaO3面上氧离子格点位置上的空穴,于是像体材料中那样产生G类反铁磁序,且氧八面体基本上没有畸变。同时由于空穴的掺入,此类超晶格是金属性的。而对n型超晶格而言,电子从LaAlO3侧转移到SrMnO3中,并不均匀地分布在Mn离子上,占据Mn的eg轨道,使得Mn离子之间通过双交换耦合,形成该体系中的半金属铁磁性。增加SrMnO3层数,每个Mn离子的磁矩之和基本保持不变,说明转移过来的电子数是个常数。在这些结果基础之上,我们的下一步工作就是研究氧空位等内禀因素对该体系电磁性质的影响,进一步探索这类体系在自旋电子学上的应用前景。
基金项目
江苏省科技厅资助项目(项目编号BK20140278)。