1. 引言
系统状态突变情况普遍存在于气候系统、经济系统、金融系统等,这样的突变往往发生在一个阈值,即临界点,此时系统突然从一种状态变化到另一种状态,这就是系统发生变化的动力系统分岔理论。最近,已有证据表明,在临床医学中存在类似的现象,如在很多复杂的疾病,类似癌症的慢性疾病的发展过程中恶化是突然的,疾病状态急剧转变。为了描述复杂疾病的潜在的动力学机制,它们的演化常常被建模为依赖于时间的非线性动力学系统,其中系统的突变被看作是分叉点上的相变,例如前列腺癌、哮喘发作和癫痫发作。我们对疾病的突然恶化期或临界转变点特别感兴趣。根据疾病的进展程度,我们将这一过程分为三个阶段:正常状态-临界状态-疾病状态。1) 在正常状态系统稳定,具有较强的回复力和鲁棒性;2) 临界状态是正常状态的临界情况,此时系统不稳定,回复力和鲁棒性弱,病人经过合理治疗可以恢复到正常状态阶段;3) 系统在疾病状态处于另一稳定状态,具有较强的回复力和鲁棒性,表示病人处于重病阶段,即便大力治疗也难以恢复到正常状态 [1] 。
对于复杂疾病而言,由于正常状态和临界状态的区别不大及疾病自身的复杂性,基于差异表达数据和其他统计指标,检测其临界信息是比较困难的但又是主要目的。通过建立网络,从动态变化视角观察复杂疾病的分子机制,对复杂疾病做出早期临床诊断以便提出及时有效的治疗方法。样本在每个时间点下的数据是高维的且有噪声,主成分分析法等传统统计学方法无法准确确定临界点。临界慢化法(critical slowing-down, CSD)为一般系统提供了检测临界点早期预警信息的方法之一,在金融,物理,生物等系统中已经成为研究热点,其应用范围愈来愈广 [2] 。CSD法要求样本具有足够多的时序列数据,且变量需要包含反映动态临界慢化的变量。但是对于生物系统样本数据很难满足上述要求。基于高通量时序列样本数据,动态网络生物标记物法(dynamical network biomarker, DNB)有效地解决了CSD法的问题 [1] 。当样本量少时,基于单样本动态网络生物标志物法(single-samples DNB, sDNB),能够准确找到临界点和生物标记物 [3] 。
2. 方法
2.1. 基于多样本动态网络生物标志物法
对于复杂疾病而言,由于正常状态和临界状态的区别不大及疾病自身的复杂性,检测其临界信息是比较困难的。基于多样本动态网络生物标记物法的提出解决了这些问题。一般系统达到临界点或分岔点时,至少有一组变量(动态网络生物标志物)与其他变量存在明显区别,即这组变量间的相关性强且极不稳定,满足以下三个条件 [1] :
1) 动态网络生物标志物中元素的标准差均值(
)大幅增加;
2) 动态网络生物标志物中元素间的皮尔逊相关系数的绝对值均值(
)大幅增加;
3) 动态网络生物标志物中元素与非动态网络生物标志物中元素间的皮尔逊相关系数的绝对值均值
减少。
引入综合变量
:
(
是一个充分小的正数)
显然,
值达到最大或突然大幅增加时即系统发生突变,达到临界状态,挑选出动态网络生物标志物。通过T-检验,FDR,fold-change,聚类,显著性分析等步骤筛选一组相关性强且极不稳定的基因并确定临界点,具体步骤请详见附录1。
2.2. 基于单样本动态网络生物标志物法
样本数据少甚至只有一个样本数据时,无法计算皮尔逊相关系数。基于单样本动态网络生物标志物法(single-samples DNB, sDNB)解决了这个问题。类似动态网络生物标志物法,此方法要求挑选出的动态网络生物标志物满足以下三个条件 [3] :
1) 动态网络生物标志物中元素的表达偏差的绝对值均值(
)大幅增加;
2) 动态网络生物标志物中元素间的皮尔逊相关系数差的绝对值均值(
)大幅增加;
3) 动态网络生物标志物中元素与非动态网络生物标志物中元素间的皮尔逊相关系数差的绝对值均值(
)大幅增加。
引入综合变量
:
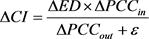 ( ε 是一个充分小的正数)
( ε 是一个充分小的正数)
显然,
值达到最大或突然大幅增加时即系统发生突变,达到临界状态,挑选出动态网络生物标志物。具体步骤请详见附录2。
3. 主要结果
3.1. 基于多样本动态网络生物标志物法有关结果
将两种方法分别应用在前列腺癌数据(GSE5345)和肝癌数据(GSE80018)两种数据,数据下载自NCBI的GEO数据库(https://www.ncbi.nlm.nih.gov/geo/)。数据具体信息及处理过程请详见附录3。
根据前列腺癌数据,利用基于多样本动态网络生物标志物法,得到综合指标CI的值在第24小时显著增加并达到峰值,动态网络生物标志物是第164类,共264个基因(含47个上游转录因子),临界点是第24小时(图1(a)),数据处理过程请详见附录3.1。另外,根据前列腺癌的动态网络生物标志物及由STRING得到的蛋白质交互网络利用Cytoscape画出疾病基因动态变化过程和翻转网络,其中发生翻转即在疾病恶
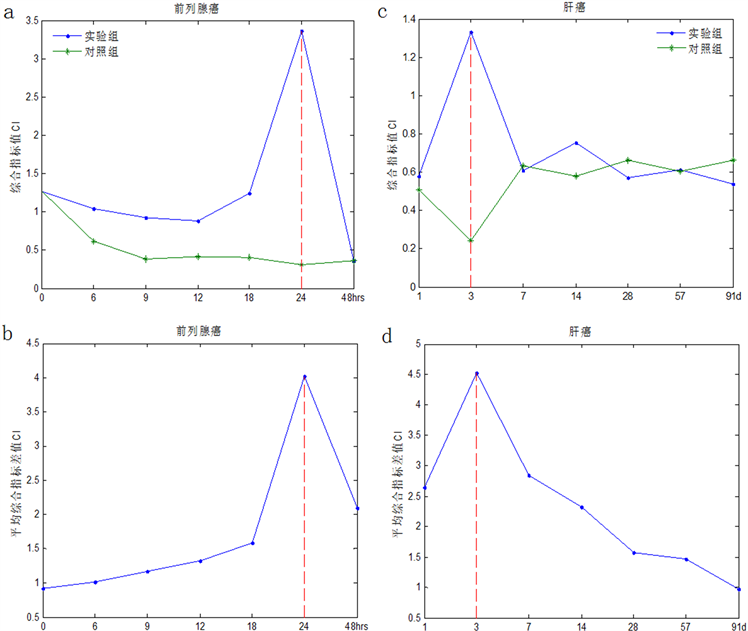
Figure 1. Line chart: early-warning signals based on multiple and single samples
图1. 多样本和单样本临界信号折线图
性突变前高表达或低表达而在恶性突变后低表达或高表达的基因有51.2%左右(图2)。结果与实验观测吻合,用合成雄激素R1881刺激人类前列腺癌细胞系LNCap 24小时后,部分RNA表达水平下降2~3倍,部分表达水平上调2~3倍,甚至3~6倍 [4] 。
类似的,根据肝癌数据,计算综合指标CI,发现其值在第3天显著增加并达到峰值,动态网络生物标志物是第164类,共139个基因(含12个上游转录因子),临界点是第3天(图1(c)),数据处理过程请详见附录3.2。结果与实验观测吻合,给小鼠喂食含0.05% [wt/vol]苯巴比妥饮用水1天后,检测到因细胞周期/有丝分裂的相关基因瞬时上调使得大部分的基因的转录水平发生变化,并且与细胞周期/干细胞调节机制的相关基因发生下调。与转录分析一致,给小鼠喂食含0.05% [wt/vol]苯巴比妥饮用水1天后,观察到包括酶CYP450和还原酶POR在内的蛋白质表达水平都发生上调。最显著的组织病理学异常是第7天开始,观察到肝细胞肥大,并在以后的时间点更加严重 [5] 。
3.2. 基于单样本动态网络生物标志物法有关结果
当样本数据少,无法利用动态网络生物标志物法确定疾病恶性突变的临界点时,基于单样本动态网络生物标志物法能够依据少量样本数据检测临界信号确定临界点,即对两个数据的单样本综合指标差值

Figure 2. Dynamically changes in the network including the DNB and overturn network during the progression of prostate cancer
图2. 前列腺癌圆盘图和翻转网络图
分别取均值确定临界点。根据前列腺癌数据,利用基于单样本动态网络生物标志物法检测4个样本都在第24小时发生突变(图1(b)),及分别有262,327,168,180个生物标记物,其中包括上游转录因子,它们两两间交集分别有103,38,37,37,84,45个基因,并对这183基因做生存分析等功能分析。根据肝癌数据,利用基于单样本动态网络生物标志物法得到5个样本都在第3天发生突变(图1(d)),及分别有190,138,186,131,170个生物标记物,它们两两间交集分别有6,9,14,19,15,9,5,28,11个基因,并对这83个基因做生存分析等功能分析,数据处理过程请详见附录4。
3.3. 功能分析
用DAVID6.8 (https://david.ncifcrf.gov/summary.jsp)对动态网络生物标志物做KEGG富集分析,前列腺癌和肝癌的动态网络生物标志物中的基因与癌症发展机制密切相关(表1,只列出部分)。基于前列腺癌数据,KEGG富集分析结果表明动态网络生物标志物与疾病的发展变化密切相关,如癌症通路、癌症转录失调、PI3K-Akt信号通路、MAPK信号通路、ErbB信号通路、Wnt信号通路等,其中大多与细胞生长、分化、增殖、凋亡,调节转录和翻译有关,Wnt信号通路异常可以导致前列腺癌。肝癌的动态网络生物标志物中的基因与代谢通路、MAPK信号通路、癌症通路、癌症转录失调等。MAPK和PI3-K/Akt通路与细胞增殖和凋亡有关,并且MAPK信号通路,ErbB信号,Wnt信号通路等通路已被证实与多种癌症的发病机理相关。KEGG富集分析表明利用动态网络生物标志物得到的结果是符合系统生物学的。
3.4. 生存分析
用SurvExpress (http://bioinformatica.mty.itesm.mx:8080/Biomatec/SurvivaX.jsp)对由基于多样本动态网络标志物法得到的生物标志物做生存分析,可知当生物体内ANXA6, CEP70, CCNA2, C-MYC, E2F-2, FES, HOXB6, IFITM1, NCOR1, POLD1, RAB11FIP1, TP53, TRAK1, UBXN11这14个基因表达异常时,前列腺癌患者存活率下降,生存时间变短(图3,请详见附录图A1);当AKAP8, COL15A1, ENO2, FUNDC2, HMGCS2, HOMER1, IHH, JUN, NDUFA1, PAM, RRAS2, TMEM106C, WIZ, WNT1这14个基因表达异常时,肝癌患者存活率下降,生存时间变短(图4,请详见附录图A2)。并且两个数据各自的动态网络标志物的生存分析的结果表明由基于多样本动态网络标志物法找到的生物标记物能够较好地反映各自癌症的临界突变信号。
由单样本得到的生物标志物,对每个单样本的生物标志物及所有两两间公共的生物标志物做生存分析,发现当AIM1L, ANXA6, CCNA2, C-MYC, E2F-2, ECT2, POLD1, RAD51AP1, TMEM19, TP53这10个基因表达异常时,前列腺癌患者存活率下降,存活时间变短(图5,请详见附录图A3);当APOE, COL15A1, HMGCS2, JUN, OLIG1, RFK, UAP1L1这7个基因表达异常时,肝癌患者存活率下降,存活时间变短(图6,请详见附录图A4)。这些分析结果证实了我们得到的动态网络生物标志物和病人的生存时间存在很强的关联,即当动态网络生物标志物所含基因表达异常时,病人的预后更差。

Table 1. Enrichment analysis by KEGG
表1. KEGG富集分析
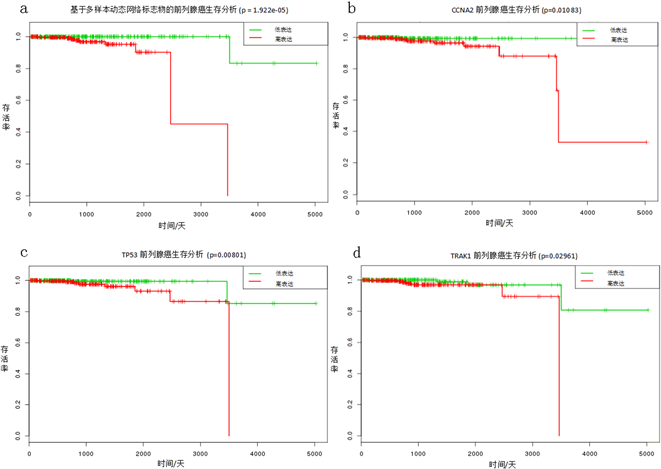
Figure 3. Survival analysis based on multi-samples of prostate cancer
图3. 基于多样本动态网络生物标志物法的前列腺癌生存分析

Figure 4. Survival analysis based on multi-samples of prostate cancer
图4. 基于多样本动态网络生物标志物法的前列腺癌生存分析

Figure 5. Survival analysis based on single-samples of prostate cancer
图5. 基于单样本动态网络生物标志物法的前列腺癌生存分析

Figure 6. survival analysis based on single-samples of liver cancer
图6. 基于单样本动态网络生物标志物法的肝癌生存分析
4 讨论
复杂疾病严重损害人类健康,我们通过检测疾病恶性突变的预警信号可以更好预防和治疗复杂疾病。虽然关键是通过检测疾病恶性突变的临界信号来采取适当的干预措施防止疾病继续恶化,但是在疾病的发展过程中,由于临界突变前系统可能没有显著变化,准确检测疾病的临界信号仍富有挑战。这也是基于传统生物标志物方法可能无法诊断疾病临界信号的原因。而动态网络生物标志物方法基于生物网络的动态性质,揭示了时序列数据在接近突变点时的临界信号,这有助于我们确定许多复杂疾病的新治疗方案的有效时间窗。
基于前列腺癌数据,利用动态网络生物标志物和生存分析等方法我们一共找到了18个基因(AIM1L, ANXA6, CEP70, CCNA2, C-MYC, E2F-2, ECT2, FES, HOXB6, IFITM1, NCOR1, POLD1, RAB11FIP1, TP53, TRAK1, UBXN11, RAD51AP1, TMEM19),当它们表达过低或过高时,会使得患者存活率下降,生存时间变短。已有研究表明这18个基因多数与细胞增殖、分裂、凋亡等有关,当它们异常表达时可能会引起细胞癌变进而诱发癌症。如C-MYC能使细胞无限增殖,促进细胞分裂,引发细胞凋亡。C-MYC与多种肿瘤发生发展有关,关于前列腺癌的大量研究表明多达30%的癌症甚至在癌前病变阶段的C-MYC基因的拷贝数已经增加了[6] [7],在前列腺肿瘤细胞中EGR过度表达可能激活C-MYC促进肿瘤的发展[8]。TP53是抑癌基因,在许多人类癌症中发生突变,TP53的错义突变、插入或缺失造成的失活突变非常常见,基因突变频率确实很高,尤其在前列腺细胞中[9]。NCOR1与多种人类恶性肿瘤相关,它在调节各种核受体以及染色体重塑方面发挥重要作用,与膀胱癌及前列腺癌的发生、发展、预后及治疗的敏感性密切相关,NCOR1在前列腺癌组织中的表达较癌旁组织明显升高[10]。
基于肝癌数据,利用动态网络生物标志物和生存分析等方法一共找到了18个基因(AKAP8, APOE, COL15A1, ENO2, FUNDC2, HMGCS2, HOMER1, IHH, JUN, NDUFA1, OLIG1, PAM, RFK, RRAS2, TMEM106C, UAP1L1, WIZ, WNT1),当它们过高或过低表达时会使得患者存活率下降,生存时间变短。这18个基因已有多数被证实与癌症有关。如COL15A1的缺少与肌肉和微血管恶化有关,与癌旁组织相比,它在肿瘤中表达显著增加[11]。ENO2是一个重要的肺癌肿瘤标志物,能作为生物标志物来帮助识别乳腺癌神经内分泌分化,与人类恶性肿瘤有关[12] [13]。HMGCS2在肝脏中的表达与肝脏癌前病变和肝癌发展有关,细胞分化时HMGCS2表达增加,是C-MYC的直接靶节点,C-MYC能抑制HMGCS2转录活性[14] [15]。C-JUN通过拮抗P53的活性来抑制TNF-α诱导的细胞凋亡,进而促进肝肿瘤的发展[16]。
附录
1. 基于多样本动态网络生物标志物法
基于以上定理和结论,对于复杂生物系统的多样本数据或高维时间序列数据,利用基于多样本动态网络生物标志物法我们可以检测生物系统复杂疾病的动态网络生物标志物(
)和早期预警信号,为疾病的临床诊断和治疗做贡献。
可以仅依据数据得到,数据具体处理过程如下:
Step 1:标准化
(1)
(2)
表示正常样本数据;
表示疾病样本数据;
分别表示正常样本数据和疾病样本数据标准化后的数据;
,
分别表示正常样本数据的均值和标准差。
Step 2:T-检验
在每个时间点,设置显著性水平
或者
对数据进行T-检验,挑选出在正常样本和疾病样本间具有显著差异的基因。
Step 3:计算伪发现率FDR
伪发现率(FDR, false discovery rate)是统计学名词,其意义是拒绝真的原假设的个数占所有被拒绝的原假设个数的比例的期望值,可以作为筛选出的差异变量的评价指标。FDR的取值可以灵活调整,通常定为0.05。设定一个显著性水平阈值
,使得FDR被限制在某一固定水平,分两步进行:首先将所有检验的
值按升序排序,即
;然后逐步后退比较
,取第一个满足条件的
,理论上可以证明在此情况下可以将FDR控制在水平
之下。
Step 4:差异表达Fold-change
利用标准差进一步筛选显著性基因。
Step 5:聚类
利用皮尔逊相关系数对基因聚类,每个类中基因间的相关性高。由以上理论知识可知,若一个时间点接近或是临界点,那么这个时间点的类中满足条件的基因即为备选
,生物系统在这个时间点达到临界状态,疾病恶化。
Step 6:显著性分析
根据
的三个指标,确定
。
计算综合指标值
,
(3)
其中,
是
中所有基因的标准差的均值,
是
中基因间皮尔逊相关系数的绝对值的均值,
是
中的基因与其他基因间皮尔逊相关系数的绝对值的均值,
是一个充分小的数,以保证整个分式有意义。
最后观察
的动态变化确定疾病恶化的早期预警信号和动态网络生物标志物。
2. 基于单样本动态网络生物标志物法
样本数据少甚至只有一个样本数据时,无法计算皮尔逊相关系数。基于单样本动态网络生物标志物法(single-sample DNB, sDNB)解决了这个问题。类似动态网络生物标志物法,此方法要求挑选出的
满足正文2.1中的三个条件具体过程如下:
Step 1:计算表达偏差
筛选基因
(4)
计算每个基因的表达偏差值,设置合适的阈值筛选基因。
Step 2:聚类
利用皮尔逊相关系数差值的绝对值对基因聚类,距离函数设为
。
Step 3:计算
假设有
个对照组,再加入一个测试样本后,计算每类基因组中的
,根据其显著性挑选显著表达的边。用Z-检验计算边的显著性值
,设定阈值以筛选边,
(5)
(6)
Step 4:计算指标值
:
 (7)
(7)
依据条件筛选基因和边,确定疾病恶化的临界点和DNB。
3. 基于多样本动态网络生物标志物方法的应用
从NCBI数据库下载两个个真实数据(http://www.ncbi.nlm.nih.gov/geo/),找到每个指针对应的基因名,没有对应基因名的指针数据忽略不计,指向同一基因名的指针对应的数据取均值,数据具体处理过程如下。
3.1. 前列腺癌
从NCBI数据库下载前列腺癌数据GSE5345。该实验数据是基于前人的实验工作得到的。简单地说,用合成雄激素R1881(实验组)和酒精(对照组)刺激人类前列腺癌细胞系LNCap,研究合成雄激素R1881对前列腺癌细胞系LNCap的基因表达的影响。每个时间点4个样本,研究人员对实验组和控制组分别在0,6,9,12,18,24,48小时(共7个时间点,表S1)时分离RNA,收集基因表达数据,一共58个样本数据。NCBI没有提供实验组在0小时处的数据,这里用对照组在0小时处的数据代替。利用微阵列数据显著性分析发现至少有3个连续时间点对雄激素刺激反应有统计学意义的变化,实验的具体过程请详见原文。合成雄激素R1881是一种口服活性蛋白同化雄性类固醇,是被广泛应用于科学研究中的雄激素受体。
基于多样本动态网路生物标志物法,该数据的具体处理过程及结果如下:
Step 1:数据预处理
此数据具有4224个原始指针,忽略没有对应基因名的指针并对指向同一基因名的指针数据取均值,最后剩下2674个基因数据。根据T-检验(显著性水平
)挑选出1004个显著表达基因映射到STRING上得到976个基因和31,496条边,用以画动态网络生物图。按照公式(1)、(2)对实验组和对照组数据标准化处理。
Table A1. Descriptions of the two datasets
表A1. 前列腺癌数据和肝癌数据
Step 2:筛选显著性基因
在每个时间点对数据进行T-检验(显著性水平
)和FDR处理(
),在每个时间点筛选出的基因数都是2674个;设置差异表达倍数值为4,在每个时间点得到的基因数为[0, 153, 139, 178, 301, 336, 74]。
Step 3:聚类
在每个时间点,利用皮尔逊相关系数对基因聚类,聚类数是40。
Step 4:计算综合指标值
依据公式(3)计算前列腺的综合指标值,确定临界点。
综合指标
的值在第24小时显著增加并达到峰值,动态网络生物标志物是第164类,共264个基因,动态网路生物标志物中的基因在12小时处综合指标值
开始骤增并在24小时处达到最大值,系统在24 h处发生突变,出现早期预警信号(图1(a)),结果与实验观测吻合。
对两类数据所得的动态生物标志物用KEGG富集分析和生存分析,发现动态生物标志物中的基因与疾病发展密切相关(表1,图S1)。
3.2. 肝癌
从NCBI数据库下载肝癌数据GSE80018。对公鼠B6C3F1分别喂食含0.05% [wt/vol]苯巴比妥(Phenobarbital, PB)饮用水和不添加PB的正常饮用水,分别作为实验组和对照组。在实验时间第1,3,7,14,28,57,91天收集基因表达数据,每个时间点5个样本(表S1),研究PB对小鼠肝脏的致癌作用。
根据基于多样本动态网路生物标志物法的理论知识,该数据的具体处理过程及结果如下:
Step 1:数据预处理
此数据具有39653个原始指针,忽略没有对应基因名的指针并对指向同一基因名的指针数据取均值,最后剩下21495个基因数据。再根据(1)、(2)对实验组和对照组数据标准化处理。
Step 2:筛选显著性基因
在每个时间点对数据进行T-检验(显著性水平
)和FDR处理 (
),筛选出的基因数为[6789, 3436, 1678, 1756, 3090, 973, 1770];设置差异表达倍数值为2.5,在每个时间点得到的基因数为[788, 204, 96, 141, 110, 80, 100]。
Step 3:聚类
对以上的到的基因根据皮尔逊相关系数聚类,每个时间点基因分为40类。
Step 4:计算指标值
根据(3)计算综合指标值,观察它的变化情况,确定临界点。
得到该数据的动态网络生物标志物是第3天处的第4类,共139个基因,包括12个上游转录因子,动态网络生物标志物中的基因在第1天的综合指标值开始骤增并在第3天处达到最大值,出现早期预警信号,即第3天是该肝癌数据的临界点(图1(c)),结果与实验观测吻合。
对两类数据所得的动态生物标志物用KEGG富集分析和生存分析,发现动态生物标志物中的基因与疾病发展密切相关(表1,图S2)。
4. 基于单样本动态网络生物标志物方法的应用
基于充足样本数据,我们可以根据正文2.1的三个条件检测复杂疾病的早期预警信息,确定疾病恶化的临界点,为临床诊断和治疗做贡献。但是出于现实原因,我们无法得到充足完整的样本数据。可基于单样本数据,根据正常样本和病人在某个时间点的数据构建网络。数据具体处理过程如下:
4.1. 前列腺癌
根据公式(4)~(7)计算各个指标值,基于前列腺癌数据,以对照组的第1~9列为对照组,对于实验组中的4个测试样本设定表达偏差
的阈值分别为3.3,2.5,2.0,1.7;层次聚类数分别是50,20,20,10;在每个时间点下的聚类数是[12, 5, 2, 14, 40, 270, 18],[63, 243, 20, 42, 150, 304, 21],[12, 5, 2, 14, 40, 270, 18],[12, 5, 2, 14, 40, 270, 18],得到动态生物网络标志物中的基因分别有262,327,168,180个(含上游转录因子),它们两两间公共基因分别有103,38,37,37,84,45个。这里把每一列数据都看做是一个单样本,是不同个体在不同时间下测得的数据,于是将得到的综合指标差值
取均值,确定病人疾病恶性突变的临界信号,根据综合指标差值变化得知该数据的临界点是第24小时(图1(b))。对这些公共基因(183个)做生存分析知基因AIM1L,ANXA6,CCNA2,C-MYC,E2F-2,ECT2,POLD1,RAD51AP1,TMEM19,TP53表达过高或过低时会使得病人的存活时间变短,存活率下降(图S3)。
4.2. 肝癌
基于肝癌数据,以对照组的第27~35列为基于单样本动态网络生物标志物方法中的对照组,对于实验组中的5个测试样本设定表达偏差
的阈值分别为3.4,3.6,2.8,3.0,3.5;层次聚类数都是30;在每个时间点下的聚类数是[169, 210, 21, 101, 118, 5, 47],[52, 162, 99, 13, 7, 2, 2],[973, 207, 1146, 346, 41, 156, 430],[523, 147, 189, 97, 117, 186, 229],[72, 174, 13, 31, 8, 88, 34]。动态生物网络标志物中的基因分别有190,138,186,131,170个,并且其中分别包括9,5,8,13,25个上游转录因子,它们两两间公共基因分别有6,9,14,19,15,9,5,28,14,17个。这里把每一列数据都看做是一个单样本,是不同个体在不同时间下测得的数据,于是将得到的综合指标差值
取均值,确定病人疾病恶性突变的临界信号,根据综合指标差值变化得知该数据的临界点是第3天(图S1(d))。对这些公共基因(82个)做生存分析可知基因OLIG1,RFK,HMGCS2,UAP1L1,COL15A1,APOE,JUN表达过高或过低时会使得病人的存活时间变短,存活率下降(图S4)。

Figure A1. Survival analysis based on multi-samples of prostate cancer
图A1. 基于多样本动态网络生物标志物法的前列腺癌生存分析

Figure A2. Survival analysis based on multi-samples of liver cancer
图A2. 基于多样本动态网络生物标志物法的肝癌生存分析
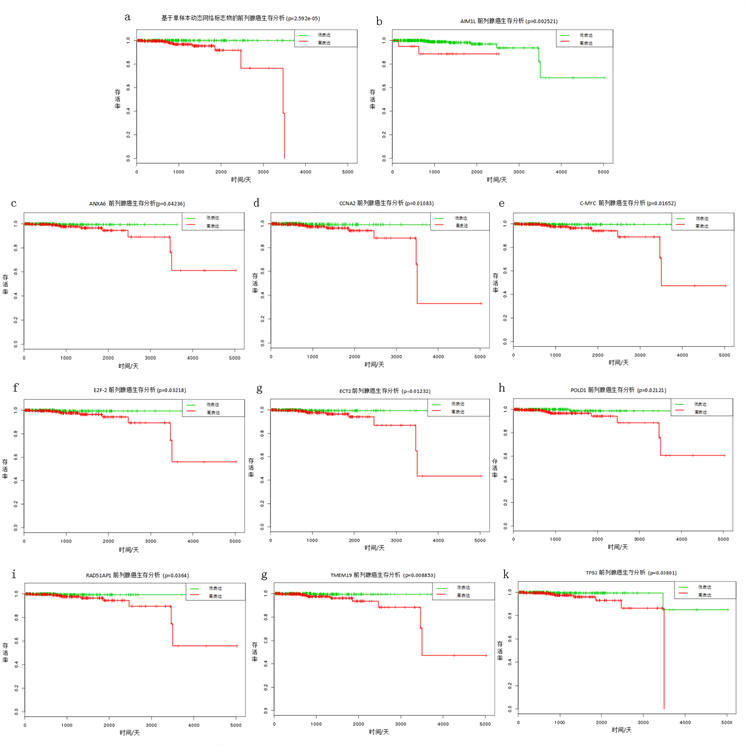
Figure A3. Survival analysis based on single-samples of prostate cancer
图A3. 基于单样本动态网络生物标志物法的前列腺癌生存分析
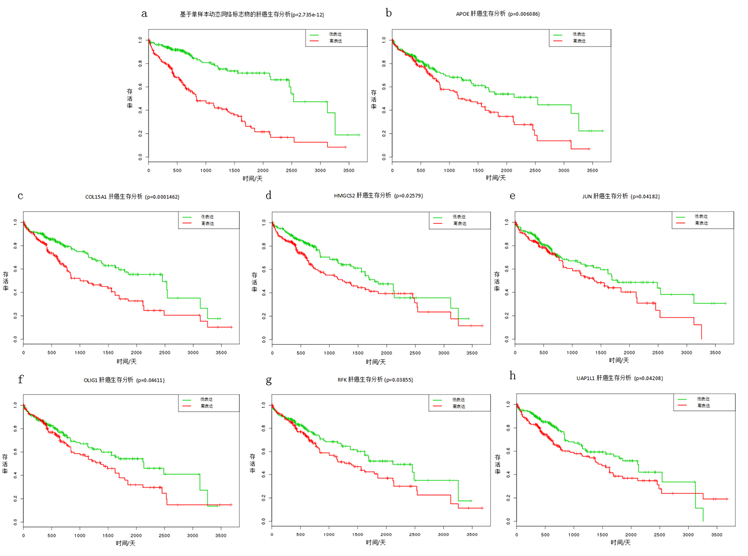
Figure A4. Survival analysis based on single-samples of liver cancer