1. 引言
淀粉是一种来源丰富、价格低廉、无毒的天然可再生资源,淀粉膜可广泛应用于可降解塑料、包装材料、生物医药等领域。作为生物医用材料使用时,其机械强度差、易酶解,故其改性一直受到研究者们的高度重视 [1] 。文献中常见的有物理改性、化学改性、生物改性和复合改性等 [2] [3] [4] 。淀粉基材料运用于生物医药领域时,评价其在体内的降解情况具有重要意义。但是文献中极少有依据人体血液中α-淀粉酶的含量(正常人血清中的淀粉酶活性范围是在21~101 U/L [5] )来评价淀粉基材料在体外酶解的情况。
本文依据三偏磷酸钠与淀粉的交联机理 [6] [7] [8] ,在淀粉糊液中引入三偏磷酸钠(STMP)和NaOH,使淀粉膜在烘干成型过程中原位交联。以pH值为7.4的磷酸盐缓冲溶液作为模拟体液,调整其酶活性浓度为100 U/L,在37℃时对不同交联程度的三偏磷酸钠(STMP)原位交联淀粉膜做酶解处理,采用3,5-二硝基水杨酸法测定酶解液中还原糖含量以评价其酶解率;研究了原位交联淀粉膜在酶解环境下的结构与性能,从而比较分析系列交联膜的酶解性能。
2. 实验
2.1. 原料
马铃薯淀粉(食品级,甘肃丰收农业科技有限公司);三偏磷酸钠(CP, Aladdin Chemistry Co. Ltd.);3,5-二硝基水杨酸(CP,国药集团化学试剂有限公司);四水合酒石酸钾钠(AR,国药集团化学试剂有限公司);α-淀粉酶(2000 U/mL,苏州宏达制酶有限公司)。
2.2. 交联马铃薯淀粉膜的制备
分别取14 g马铃薯淀粉,2.8 g甘油,0.27 g氢氧化钠,150 mL去离子水加入到500 mL三口烧瓶中。在75℃下搅拌预糊化30 min,加热至95℃后继续搅拌1 h,搅拌速度保持150 rpm,自然冷却至25℃,之后分别加入不同质量分数(0 wt%, 1 wt%, 3 wt%, 6 wt%, 9 wt%, 15 wt%)的交联剂,搅拌均匀10 min。然后将糊液在真空下脱泡处理,刮膜,制备淀粉膜(Potato Starch Film,简写为PSF)及不同交联剂含量的交联淀粉膜(Cross-linked Potato Starch Film,简写为CPSF)。置于50℃、70%RH的恒温恒湿箱中干燥24h,取出揭膜,制得编号分别为PSF、CPSF1、CPSF3、CPSF6、CPSF9、CPSF15的交联膜。
2.3. 交联马铃薯淀粉膜的平衡溶胀率测试
将膜材料裁剪成20 mm × 20 mm的样品,溶胀测试前,先将膜置于鼓风烘箱中50℃干燥至恒重W0。然后将干燥后的膜浸渍于盛有蒸馏水的培养皿中,24 h后用镊子小心取出膜,快速用滤纸擦干膜表面的水分,称重W1。平衡溶胀率ESR (Equilibrium Swelling Ratio)定义为膜充分溶胀24 h后的质量变化量。交联淀粉膜的平衡溶胀率(ESR)按式(1)计算。
(1)
式(1)中:W0代表膜干燥后的质量,g;W1代表膜充分溶胀24 h后的质量,g。
2.4. 交联马铃薯淀粉膜的酶解试验
将交联淀粉膜裁成2 cm × 2 cm大小,称重W0,置于无酶的磷酸盐缓冲溶液中浸泡以去除杂质,再置于含有一定酶浓度的磷酸盐缓冲溶液中,在37℃下的往复式水浴恒温振荡器中进行酶解处理,每隔一段时间取1 mL降解液的上清液,待测。交联淀粉膜的降解率按式(2)计算。
(2)
式(2)中:c代表降解液中还原糖的浓度,mg/mL;V代表降解液的体积,mL;W0代表交联膜的初始质量,mg。
2.5. 交联马铃薯淀粉膜湿强测定
交联淀粉膜的湿态强度较低,万能材料试验机的夹具会把膜材料夹碎,本实验中模拟万能试验机的原理自制了一台粗略测定抗拉强度的简易装置,如图1所示:
测试步骤:将交联淀粉膜裁成1.5 cm × 3 cm大小,置于PBS中充分溶胀后,用上下夹具固定湿膜,打开调节阀,使水缓慢沿水槽壁滴入水槽。膜断裂时,关闭开关,称得下方挂钩和水槽总重W2(g)。每个样品重复5次,取平均值。拉伸强度(Tensile Strength, TS)按下式(3)计算 [9] 。
(3)
式(3)中:W1代表膜的质量,g;W2代表挂钩和水槽的总质量,g;h代表膜的宽度平均值,mm;d代表厚度平均值,mm。
2.6. 降解液中多糖含量的测定
为了定量分析淀粉基材料的降解情况,常通过测定降解液中的糖含量来反应材料的降解情况。在多糖的定量分析中,常见的测定方法有苯酚-硫酸法、蒽酮-硫酸法和3,5-二硝基水杨酸法(简称DNS法)等 [10] 。但前两种方法测定步骤繁琐,且苯酚极易氧化,硫酸具有严重的腐蚀性,给实验操作带来极大不便。3,5-二硝基水杨酸法具有操作简便、快速、灵敏度高、杂质干扰小等优点,近年来逐渐被应用于植物药、中成药、海洋生物药等药物中多糖含量的测定 [11] [12] 。
1) 标准曲线的绘制
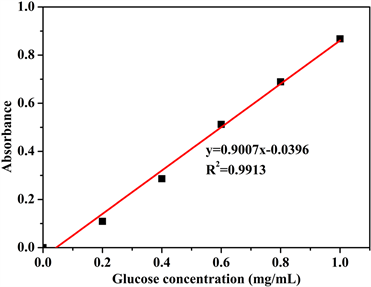
分别取葡萄糖标准溶液0、0.2、0.4、0.6、0.8、1.0 mL于15 mL试管中,编号分别为1、2、3、4、5、6,用蒸馏水补足定容至1.0 mL,分别加入DNS试剂2 mL,沸水浴加热5 min,迅速用流水冷却,再用蒸馏水补足至15 mL。以1号试管为空白对照,540 nm波长下测定各管显色液的吸光度。绘制吸光度-葡糖糖浓度曲线,如图2所示。
用最小二乘法进行线性回归,得到吸光度与葡糖糖浓度的关系式为:y = 0.9007x − 0.0396,R2 = 0.9913,
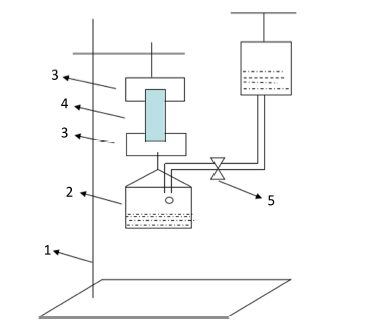
Figure 1. Test device of wet strength of films
图1. 湿强度测试装置(1,铁架台;2,水槽;3,夹具;4;样品;5,调节阀)
 (a)
(a) 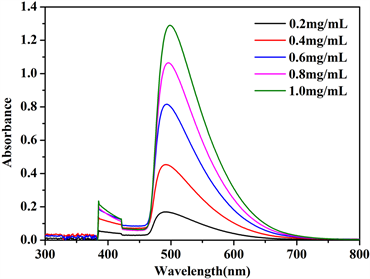 (b)
(b)
Figure 2. The standard curves of absorption and glucose concentration: (a) UV-vis absorption spectra of glucose colorimetric solutions with different concentrations; (b) standard curves
图2. 吸光度-葡萄糖浓度标准曲线:(a) 紫外可见吸收光谱;(b) 标准曲线
吸光度与葡萄糖的浓度在测量范围内呈良好的线性关系。
2) 样品中多糖含量测定
采用Lamda35型紫外-可见分光光度仪测量标准曲线样品及降解液样品的紫外可见吸收光谱。
2.7. X射线衍射分析
将未交联及交联淀粉膜烘干并研成粉末,测试淀粉膜的结晶性能,测试条件:铜靶,管压40 kV,管流200 mA,扫描范围5˚~60˚。
2.8. 傅里叶红外光谱分析
将淀粉膜置于去离子水中充分溶胀以去除反应后的杂质,然后放入真空烘箱中烘干,研成粉末,采用KBr压片法测试,扫描范围4000~400 cm−1,扫描次数32次,频率4 cm−1。
2.9. 扫描电子显微镜分析
将膜样品裁剪成长条状于液氮中脆断后,在断面喷金,另取一些表面喷金,在电镜下观察膜的表面和断面形貌。
3. 结果与讨论
3.1. 交联马铃薯淀粉膜的FTIR分析及其平衡溶胀率
采用FTIR对比分析淀粉膜交联前后的化学结构变化,并通过平衡溶胀率定性地描述其交联程度,分别见图3和表1。
由图3可见,934 cm−1、860 cm−1、763 cm−1处为淀粉的特征峰,即为α-D-吡喃葡萄糖的骨架振动峰,3400 cm−1附近的吸收峰为-OH的伸缩振动峰,1640 cm−1处的吸收峰为淀粉分子内结合水的-OH的弯曲振动峰,2931 cm−1处的吸收峰为饱和碳氢键C-H的伸缩振动峰,1154 cm−1处的吸收峰为C-O-C基团上的C-O伸缩振动峰,1081 cm−1、1024 cm−1处的吸收峰为C-O-H基团上的C-O伸缩振动峰 [13] [14] [15] [16] 。1239 cm−1处的峰可归属于P=O的伸缩振动峰,1206 cm−1处的峰可归属于P-O-C的伸缩振动峰 [17] ,并且与PSF相比:CPSF在这两处的吸收峰均有所增强,这可能是由于交联反应引入的磷酸酯基团使得P=O
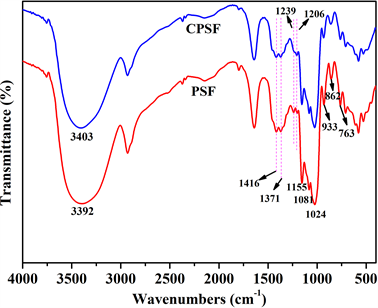
Figure 3. FTIR spectra of starch films
图3. 淀粉膜的FTIR谱图
和P-O-C的伸缩振动峰增强;CPSF的-OH的伸缩振动峰变窄,且向高波数移动,这说明-OH的振动种类减少,-OH被交联键取代,即发生了交联反应 [18] 。以上这些红外谱图上细微的变化均为STMP与淀粉分子的交联反应生成的磷酸酯基团提供了有利的证据。依据三偏磷酸钠与淀粉的交联机理,在淀粉糊液中引入三偏磷酸钠(STMP)和NaOH,使淀粉膜在烘干成型过程中原位交联。如图4所示,STMP分子中的P-O-H与不同淀粉分子链上的伯羟基发生反应而形成交联,使两个或两个以上的淀粉分子之间通过化学键键接在一起,呈现多维空间交联网络结构 [19] [20] 。
溶胀率一方面可反映膜材料各组分对膜吸水溶胀性能的贡献;另一方面溶胀率也可以表征膜材料中亲水基团的情况,亲水基团越多或者基团亲水性越好,则膜材料溶胀性就越好。本章制备的交联淀粉膜交联程度越高,膜材料中亲水基团减少,且交联形成的三维网络结构更加致密,导致膜的溶胀率下降。因此,膜的溶胀率在一定程度上可以反映出膜的交联程度。不同交联程度样品的平衡溶胀率如表1所示。由表1可见,随着交联剂STMP含量的增加,对应膜的平衡溶胀率逐渐降低,表明淀粉膜体系中亲水性基团逐渐减少,交联程度也逐渐增加,空间网络结构愈加致密。
3.2. 交联马铃薯淀粉膜在酶解过程中的形态与力学性能的变化
体内服役过程中膜的保形能力及其湿态环境下力学性能如何是决定其服役行为的关键。为此,本文对比分析了不同交联程度的交联淀粉膜在相同条件下酶解一定时间后的形态和力学性能变化,如图5所示。由图5可知,在酶解12 h后,未交联的淀粉膜PSF和交联程度较低的交联膜CPSF1和CPSF3已完全降解或仅剩少量的淀粉膜碎片,降解液呈澄清透明状,说明酶解过程中固体膜中的淀粉链被降解成低分子量物质而水溶;而CPSF6在酶解12 h后仍能保持完整的膜的外形,CPSF9在酶解4 d后,仍具有一定的力学强度,当交联程度最大的CPSF15,其在酶解7 d后膜的外形仍能保持,目视未观察到明显的形貌变化,并且在一定的拉伸作用下不破碎,仍保持较好的力学强度。以上现象说明随着交联程度的增大,交联膜的耐酶解能力明显提高,可在高α-淀粉酶浓度下较长时间保持力学性能而具有一定的应用价值。
进一步采用自制的湿强测试装置比较不同交联程度淀粉膜的湿态强度及其在酶解过程的变化,其

Table 1. Equilibrium swelling ratio of CPSF with different contents of STMP
表1. 交联淀粉膜的平衡溶胀率
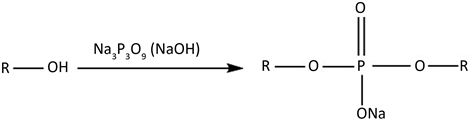
Figure 4. Proposed mechanism of the cross-linking reaction of starch with STMP
图4. 淀粉与STMP的交联反应机理

Figure 5. Photos of films with different degradation time
图5. 不同交联剂含量的交联淀粉膜的形态随酶解时间的变化
结果见图6。由图6可见:1) STMP交联能够较大地提高淀粉膜的湿强,并且改善效果随交联程度的增大而加强,CPSF15的湿强可达近60 kPa,差不多是低交联淀粉膜CPSF1的3倍,而未交联淀粉膜PSF溶胀后不能保形;2) 则其交联膜的湿强随酶解时间的延长而降低,这是由于α-淀粉酶作用于淀粉时,从内部随机破坏淀粉分子中的α-1,4-糖苷键,使其力学性能降低 [21] 。在误差允许范围内,交联剂含量越大,湿强越大,降解周期也越长,因为交联淀粉膜中酶作用位点减少,酶解速率降低。
3.3. 淀粉膜的降解率测定
为了定量分析淀粉膜的降解程度,采用DNS法显色并测定酶解不同时间的酶解液中还原糖的含量,图7为不同交联程度交联淀粉膜的酶解液显色照片。由图7可见,未交联PSF随着酶解时间的延长,酶解液中的还原糖(葡萄糖、麦芽糖等)含量不断增加[22],显色后溶液的颜色呈棕色并随着葡萄糖含量的增大而加深。而随着膜交联程度的加大,酶解液呈棕红色变深的时间延长,尤其当交联剂含量增加到15 wt%时,CPSF15酶解7 d后,酶解液显色后仍然未见明显的棕红色,说明CPSF15在淀粉酶的作用下7 d后,仍未发生明显的降解,这是因为该膜的平衡溶胀率相对最小,交联程度高,形成了致密的交联网络结构,即使在酶解液体系的溶胀环境中,淀粉分子链间也保持足够紧密的距离,从而使得淀粉酶分子难以与淀粉分子链中的1,4-糖苷键接触而阻碍酶解反应的发生。
采用紫外-可见光吸收光谱定量分析酶解液中小分子还原糖的含量并进一步得到降解程度与酶解时间的关系曲线(图8)。由图8可见,在交联程度较低时,由酶解生成的小分子还原糖浓度所反映出的膜的降解进程,明显分为两个阶段:酶解初期的快速降解阶段和后期的近匀速降解阶段,这表明在淀粉膜中存在可及性不同的区域:1) 酶分子高可及区,该区域在酶解液体系中可溶胀而使得淀粉分子链间的距离
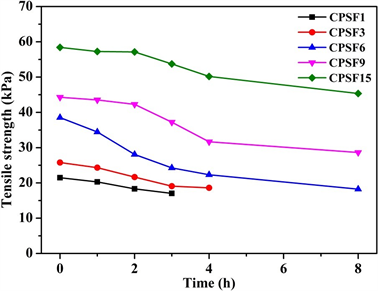
Figure 6. Wet strength of films with different degradation time
图6. 交联膜的湿强随降解时间的变化
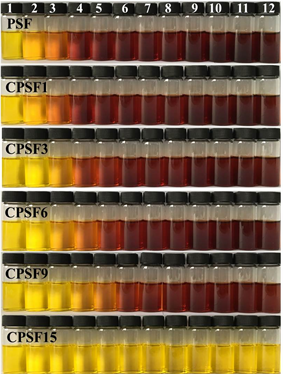
Figure 7. Photos of colorimetric solutions of degraded films, degradation time: (1) 1 h; (2) 2 h; (3) 4 h; (4) 8 h; (5) 12 h; (6) 24 h; (7) 2 d; (8) 3 d; (9) 4 d; (10) 5 d; (11) 6 d; (12) 7 d
图7. 不同交联剂含量的交联淀粉膜的降解液显色照片,降解时间:(1) 1 h; (2) 2 h; (3) 4 h; (4) 8 h; (5) 12 h; (6) 24 h; (7) 2 d; (8) 3 d; (9) 4 d; (10) 5 d; (11) 6 d; (12) 7 d
变大,从而使得淀粉酶分子可扩散进入发生酶解反应,酶解初期的酶解反应主要发生在该区域;2) 酶分子低可及区,该区域分子链排列紧密(因结晶和/或交联),溶胀程度小,淀粉酶分子较难扩散进入该区域,酶解反应发生在该区域表面,由表及里逐步反应,后期的近匀速酶解阶段的酶解反应发生在该区域。而当膜的交联程度达到一定值时,如CPSF15,因所有分子链均被充分、高度交联,溶胀程度低(由表1可见),酶解反应仅发生在膜的表面,故所能测得的小分子还原糖浓度很低。
淀粉膜中高可及区和低可及区的存在可以由其XRD证实(见图9),未交联淀粉膜PSF在2θ 20˚附近
Figure 8. The relationship curves of degradation ratio and time while the films was treated by α-amylase solution
图8. 不同交联程度淀粉膜的降解程度与酶解时间的关系曲线
出现了明显的淀粉分子的结晶峰 [23] ,这说明反应初期酶解反应发生在膜的非晶区,而后期发生在结晶区,而交联淀粉膜CPSF9的XRD图谱表明该固态膜中淀粉分子链基本处于无定形状态(分子链有序化程度低),其原因是干法成型时同时发生的交联反应抑制了淀粉分子链的运动和有序化堆砌而形成高度有序化的结晶区,随着交联程度的增大,因交联而导致的分子链上亲水基团的减少以及分子链间更为致密,使得无定形区的可及性不断降低,直至CPSF15的交联程度时,无定形区也处于不可及状态。由上述讨论可知,淀粉膜的交联程度对酶解反应影响很大,交联反应使淀粉分子中酶作用的位点减少,阻碍了淀粉膜的酶解反应 [24] 。
3.4. 酶解对交联淀粉膜化学结构和形貌的影响
采用FTIR (图10)和SEM (图11)表征分析了交联淀粉膜酶解前后的化学结构和微观形貌变化。
由图10可见,交联膜酶解前后的谱图FTIR基本相似,在波数和峰强方面没有明显变化,这是因为
α-淀粉酶对淀粉分子的作用是无差别切断α-1,4糖苷键,产生不同聚合度的低聚糖,故红外谱图差别不大。
图11为PSF及CPSF15酶解前后的表面及断面的扫描电镜照片。由图9(a)(b)(e)(f)可见,未酶解PSF
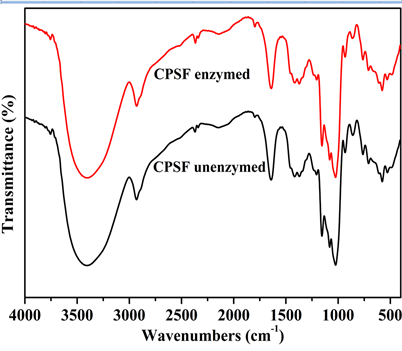
Figure 10. FTIR spectra of CPSF unenzymed and enzymed
图10. 交联膜酶解前后的傅里叶变换红外光谱图
和CPSF15的表面及断面呈现光滑致密结构,无明显的缺陷。PSF经酶解2 h后,表面出现许多的孔状或带状的结构,断面基本为连续致密的状态,仅在靠近PSF表面处才出现一些带状结构,这是由于相比CPSF15,PSF酶作用位点多,与α-淀粉膜可快速发生反应,酶解掉的淀粉膜溶于降解液中,导致PSF表面出现明显的多孔网络形貌。PSF酶解前的厚度大约为150 μm,酶解2 h后厚度大约为55 μm,说明PSF经酶解2 h后发生了很大程度的降解。CPSF15经酶解4 d后,表面有少数凹凸不平的结构,并无明显的缺陷,断面基本为连续致密的状态,CPSF15酶解前厚度大约为175 μm,酶解4 d后的CPSF15厚度大约为160 μm,说明CPSF15经酶解4 d后发生了一定程度的降解,且降解率明显比PSF低很多。从SEM分析可以看出,PSF和CPSF15的酶解主要是从膜的表面开始。
CPSF的耐酶解性能较PSF明显提高。原因可能为:交联反应后,淀粉分子内羟基数量减少,减弱了膜材料的水合能力,导致淀粉分子与α-淀粉酶分子的碰撞机会减小;另外,交联反应使得材料结构更加致密,这阻碍了α-淀粉酶分子渗入淀粉分子间。因此,交联后的淀粉膜具有较强的耐酶解性能,且可以通过改变交联程度来调控CPSF的酶解周期。
4. 结论
1) 交联剂含量为0~15 wt%时,随着交联剂含量的增加,交联膜的平衡溶胀率减小,即交联程度增大。随着交联程度的增大,交联膜的耐酶解能力不断增强,7 d内PSF降解率达到55.0%,而CPSF15的降解率仅为1.6%。
2) 交联淀粉膜酶解过程中,随交联剂含量越大,湿强越大,降解周期也越长。
3) 由淀粉膜酶解前后的SEM分析可知,PBS中淀粉膜在α-淀粉酶作用下的酶解过程是由表及里的,对膜材料致密部分的降解缓慢。