1. 前言
2003年,Anastassiades等 [1] 开发了一种快速(quick)、简单(easy)、便宜(cheap)、高效(effective)、耐用(rugged)和安全(safe)的农产品中农药多残留样品前处理方法,并用首字母缩写将此种方法命名为QuEChERS。自其发布以来,因其简化了以前繁杂的萃取步骤并扩大了所萃取农药残留的范围,因此,在农药残留领域得到了大量的应用。
喹诺酮类(Quinolones),又称吡酮酸类或吡啶酮酸类,具有广谱抗菌、价格低廉的特点 [2] ,因此该类药物广泛应用于畜禽养殖环节,然而其使用也引发出一系列的问题。目前,喹诺酮类药物残留检测方法的不确定度评价主要为高效液相色谱法 [3] 、高效液相色谱串联质谱法等 [4] [5] 。
测量不确定度是测量结果可信程度的定量表达 [6] ,其数值反映了测量结果质量,并直接关系到检验结果的合格与否。本实验参照JJF 1059.1-2012《测量不确定度评定与表示》 [6] 和JJF 1135-2005《化学分析测量不确定度评定》 [7] ,通过QuEChERS方法包提供的使用说明书,评定鸡肉中环丙沙星、恩诺沙星和氧氟沙星3种喹诺酮类药物残留检测的不确定度 [8] [9] [10] ,了解整个检测过程中不确定度的主要来源,以便实验操作者对其加以重视,从而确保检测结果的可靠性,为实验室的质量控制提供重要依据。
2. 材料与方法
2.1. 材料与试剂
鸡肉(市售);盐酸环丙沙星(94%)、恩诺沙星(98.5%)、氧氟沙星(99%),德国DR.E公司;恩诺沙星D5盐酸盐(99%)、环丙沙星D8盐酸盐(99.8%)、氧氟沙星D3盐酸盐(99.4%),德国Witega公司;乙腈(色谱纯),美国fisher公司;甲酸(色谱纯),上海麦克林生化科技有限公司;QuEChERS方法包,日本岛津公司;GHP微孔滤器(0.22 μm),美国waters公司。
2.2. 仪器设备
5500型高效液相色谱–串联质谱仪(配ESI;离子源):美国AB公司;XS205电子天平:瑞士METTLER TOLEDO公司;3-18 K高速离心机:德国Sigma 公司;涡旋振荡器:德国IKA公司。
2.3. 方法
2.3.1. 样品前处理方法
按照日本岛津公司提供的方法,准确称取5.00 g待测试样品,加入0.1 mg/L的同位素内标50 μL,加入5%甲酸乙腈溶液5.0 mL,涡旋1 min,加入WondaPak专用提取包,涡旋1 min,12,000 rpm离心5 min,取上清液1 mL于15 mL离心管中,涡旋1 min,12,000 rpm离心5 min,取上清液过GHP微孔滤器,进LC-MS/MS分析检测。
2.3.2. LC-MS/MS测定条件
色谱柱:C18,50 × 2.1 mm,3 μm;流动相:A相:0.1%甲酸乙腈溶液;B相:0.1%甲酸水溶液;梯度洗脱(见表1);柱温 40℃;进样量为1 μL;离子源:电喷雾ESI,正离子;扫描方式:多反应监测MRM;帘气:35.0;离子源温度:550℃;Gas 1:55.0;Gas 2:55.0;监测离子对及对应质谱参数(见表2)。

Table 1. The mobile phase gradient of liquid chromatography for the separation of Quinolones
表1. 分离喹诺酮类药物的液相色谱流动相梯度
Table 2. The acquisition parameters of UPLC-MS/MS for analysis of Quinolones and their isotopic labels
表2. 喹诺酮类药物及其同位素标记物质谱参数
2.4. 数学模型的建立
目标物残留量X计算方法为:
式中:X——样品中目标物残留量(μg/kg);
C——样液中目标物的浓度(ng/mL);
V——定容体积(mL);
m——样液所代表样品的质量(g)。
3. 结果与分析
3.1. 不确定度来源分
通过对样品中喹诺酮类药物残留量检测过程进行分析,发现引入的不确定分量主要有图1所示的不确定度来源。

Figure 1. Uncertainty source for determination of Quinolones in commercialized samples of chicken with UPLC-MS/MS
图1. UPLC-MS/MS测定鸡肉中喹诺酮类药物残留量的不确定度来源
3.2. 不确定度的评定
3.2.1. 目标物浓度(C)引入的不确定度
1) 储备液配制过程产生的不确定度u(C1)
分别准确称取11.81 mg盐酸环丙沙星(94%)、10.15 mg恩诺沙星(98.5%)、10.10 mg氧氟沙星(99%),11.26 mg恩诺沙星D5盐酸盐(99%, 0.2%)、11.20 mg环丙沙星D8盐酸盐(99.8%, 0.2%)、10.06 mg氧氟沙星D3盐酸盐(99.4%, 0.2%)于10 mL容量瓶中,用甲醇溶解并定容至,得到1000 mg/L的各标准储备液。该过程引入的不确定度见表3。
Table 3. Uncertainty resulting from reserve-fluid preparation
表3. 储备液配制过程引入的不确定度
2) 储备液稀释过程产生的不确定度u(C2)
用200 μL移液器移取储备液100 μL于10 mL容量瓶(A级)中,用乙腈定容,摇匀,得到10 mg/L的混合标准溶液,再移取100 μL该标准溶液于10 mL容量瓶(A级)中,用0.1%甲酸乙腈溶液定容,得到0.1 mg/L的标准工作液,再移取100μL浓度为0.1 mg/L的标准工作液于10 mL容量瓶(A级)中,用0.1%甲酸乙腈溶液定容,得到0.01 mg/L的标准工作液。
按照均匀分布处理,该过程引入的不确定度见表4。
Table 4. Uncertainty resulting from stock solution dilution
表4. 储备液稀释过程产生的不确定度
则储备液稀释过程中引入的不确定度为:
。
3) 标准系列溶液配制过程产生的不确定度u(C3)
分别移取0.1 mg/L的喹诺酮类药物混合标准溶液200、100、50、20 μL,0.01 mg/L的喹诺酮类药物混合标准溶液100、50、20 μL,各加入同位素标记混合标准溶液100 μL,分别加入流动相700、800、850、880、800、850、880 μL,得到制得20.0、10.0、5.0、2.0、1.0、0.5、0.2 ng/mL各对照溶液,从低浓度到高浓度逐一测定,每一浓度进样2次,按所得目标物与其同位素标记物峰面积之比与相应的对照溶液浓度作标准曲线,并依此计算回归方程及相关系数。
移液器刻度误差参考JJG 646-2006《移液器检定规程》 [12] 均匀分布处理,系列标准溶液配制过程中,由器具及温度波动带来的不确定度见表5。
Table 5. Uncertainty resulting from standard solution preparation
表5. 系列标准溶液配制过程产生的不确定度
则由系列标准溶液配制过程产生的相对标准不确定度为:
。
4) 标准曲线拟合产生的不确定度u(C4)
取2.2.1.3中配制的7个标准系列浓度,从低浓度到高浓度测定,每一浓度进样2针,按所得目标物与其同位素标记物峰面积之比与相应的对照溶液浓度作标准曲线,计算得到回归方程及相关系数,其中氧氟沙星
;环丙沙星
;恩诺沙星
。对阳性样品中各目标化合物浓度C0重复检测3次,检测结果为氧氟沙星5.011、4.983、5.007 ng/g;环丙沙星4.997、4.967、5.014 ng/g;恩诺沙星5.021、5.014、4.982 ng/g。标准曲线拟合产生的不确定度按照(1) (2)计算。
 (1)
(1)
(2)
n1为同一溶液测量次数;n2为标准溶液测量次数;Xi为标准溶液理论值;
为标准溶液浓度平均值;Y为标准溶液峰面积与内标峰面积的比值;b为标曲斜率;c为标曲截距。
由(1)、(2)算出标准曲线拟合产生的不确定度分别为:氧氟沙星
,环丙沙星
,恩诺沙星
。
根据
可计算出被测物质量浓度C引入的不确定度,相关量值见表6。
Table 6. Calculation of uncertainty resulting from analyte concentration
表6. 被测物质量浓度C引入的不确定度
3.2.2. 样品称量引入的不确定度u(m)
称取5.00 g鸡肉样品,所用天平校准证书中天平的最大允许误差为±0.02 g,按照均匀分布进行计算,引入的不确定度
,则相对不确定度
。
3.2.3. 体积量取产生的不确定度u(V)
1) 定容容量产生的不确定度u(V1)
加入5%的甲酸乙腈溶液5.0 mL,移液器体积允许误差为±0.0091 mL,按均匀分布进行计算,
;由温度变化引起的乙腈容积变化为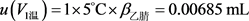 ,同样,
。则定容时产生的不确定度为:
,相对不确定度为
。
,同样,
。则定容时产生的不确定度为:
,相对不确定度为
。
2) 仪器进样体积产生的不确定度 u(V2)
LC-MS/MS进样针容积的相对标准偏差为±1%,则其相对不确定度
。
由1)和2)看出,体积量取引入的相对不确定度为:
。
3.2.4. 加入内标引入的不确定度u(V内)
由于样品与对照使用同一内标溶液,内标物的称量和稀释定容所产生的不确定度在计算过程中抵消,则对目标物含量测定有影响的不确定因素主要是内标溶液的移取引入的 [10] 。
测定过程中使用200 μL移液器,最大允差为±0.0012 mL,按照三角分布计算,移液器产生的不确定度为:
。试验温度与校正温度不同也会引入不确定度,按照均匀分布,温度差异产生的不确定度为:
。
则供试品中加入内标量引入的不确定度为:
。
相对不确定度为:
。
同理,对照品中加入内标量引入的相对不确定度
。
3.2.5. 回收率引入的不确定度u(R)
在空白鸡肉试样中添加1 μg/kg的环丙沙星、氧氟沙星、恩诺沙星混合标准溶液,平行测定10次。将平均回收率
,标准偏差S(R)分别代入公式
和
,得到由回收率带来的不确定度以及相对不确定度,测定结果见表7。
Table 7. Results of determination of Ofloxacin, Ciprofloxacin and Enrofloxacin in commercialized samples of chicken
表7. 鸡肉中氧氟沙星、环丙沙星和恩诺沙星测定结果
3.2.6. 测量重复性引入的不确定度
按照3.2.5的方法,将平均含量
,标准偏差
结果分别代入公式
和
,计算得到测量重复性引入的不确定度和相对不确定度,如表7所示。
3.3. 合成不确定度
综上各不确定度分量,根据公式
,计算可得氧氟沙星、环丙沙星和恩诺沙星的合成不确定度,结果见表8。
Table 8. Uncertainty evaluation for the determination of three Quinolones
表8. 三种喹诺酮类药物测定不确定度评定结果
3.4. 扩展不确定度
依据JJF 1135-2005,对于大多数测量采用包含因子k = 2衡量,则3种喹诺酮类药物含量的扩展不确定度
,由此得到超高效液相色谱–串联质谱法测定鸡肉中3种喹诺酮类药物含量的结果,见表8。
4. 结论
通过对超高效液相色谱–串联质谱法对鸡肉中3种喹诺酮类药物含量检测不确定度各分量的评定,发现该检测方法中,标准溶液的配制和标准曲线的拟合所产生的不确定最大,其次为测量重复性和回收率产生的不确定度分量。因此,在实际操作过程中,可通过增加混合标准系列溶液的测定次数,增加平行样品的测定次数,并定期对实验过程所涉及的仪器进行检定校准,提高操作人员的检测水平,来减小测量不确定度,从而保证检测结果的准确性。