1. 引言
反义核酸 [1] 是指能够与靶RNA (多为mRNA)碱基互补配对,引起基因沉默的寡聚DNA片段。反义核酸通过Watson-Crick碱基配对规则与靶标mRNA结合形成RNA-DNA杂交双链 [2] ,RNase H酶能够识别这种杂交双链,并特异性裂解mRNA链,达到抑制或封闭靶标mRNA表达的目的 [3] 。由于反义核酸能靶向性调控基因表达,因此被看作是一类有前景的药物。天然的反义核酸成药主要有这样几大障碍:一是难以进入细胞 [4] ,二是易被核酸酶降解 [5] ,三是可能引起潜在的免疫刺激反应 [6] 。化学修饰是改善反义核酸成药性的重要手段。三氟甲基是已知脂溶性最强的基团,在反义核酸链中引入三氟甲基基团有可能明显改善反义核酸的细胞通透性和抗核酸酶稳定性 [7] 。在本文的研究中,按照如图1所示的合成路线成功合成了C4'-CF3-α-L-脱氧胸苷亚磷酰胺单体,为后续通过固相合成方法的将其引入到反义核酸链中,研究其生化性质和成药性质打下了基础。
2. 实验部分
2.1. 实验药品与仪器
2.1.1. 实验药品
原料(S)-1,3-二(苄氧基)-4-(1,3-二噻烷-2-基)丁-2-酮通过已知路线 [8] 自己合成制备。CH2Cl2,石油醚,乙酸乙酯,无水甲醇,吡啶,NaCl,NaHCO3,MgSO4,NaOH,Na2S2O3,以上从天津市河东区广达服务部购买;醋酸酐,从天津市化学试剂供销公司购买;乙腈(99.9%超干溶剂,带分子筛),2-碘酰基苯甲酸,草酰氯,二甲基亚砜,硅藻土,以上从百灵威公司购买;胸腺嘧啶,从Sharing Technbologies Co.,Let购买;三乙胺,四氢呋喃,从北京华威锐科化工有限公司购买;Dess-Martin氧化剂,CF3SiMe3,氯化钯,N-溴代琥珀酰亚胺,三甲硅烷基三氟甲磺酸酯,4,4'-双甲氧基-三苯甲基-氯,N,N-二异丙基乙胺,以上从北京伊诺凯科技有限公司购买;四正丁基氟化铵,从希恩思奥普德科技有限公司购买;N,O-双(三甲基甲硅烷基)-乙酰胺,从塞墨飞实验室仪器销售中心购买;2-氰乙基N,N-二异丙基-氯代亚磷酰胺,从芜湖华仁科技有限公司购买;氢气,氩气从百思达气体有限公司购买;柱层析硅胶为青岛海洋公司生产的300~400目硅胶。
2.1.2. 实验仪器
核磁由Bruker公司AVANCE 400超导核磁共振谱仪测定;液相分析在waters公司的2695高效液相色谱分析仪上进行;质谱由waters公司的Xevo G2-XS Q-TOF质谱仪测定。
2.2. 实验方法
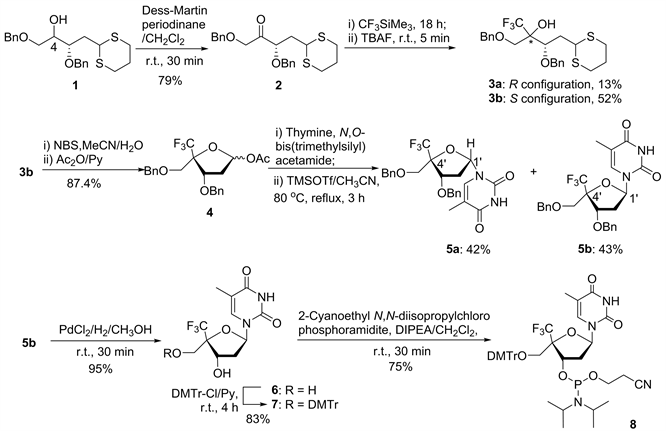
Figure 1. Synthetic route of C4'-CF3-α-L-deoxythymidine phosphoramidite
图1. C4'-CF3-α-L-脱氧胸苷亚磷酰胺单体的合成路线图
化合物2的合成。室温下将化合物1 1 g (2.7 mmol, 1 eq)溶于11 mL无水CH2Cl2,加入Dess-Martin 1.38 g (3.2 mmol, 1.2 eq)室温反应1 h,TLC监测反应结束后,加入20 mL (1:4)的饱和Na2S2O3溶液和饱和NaHCO3溶液萃灭反应,CH2Cl2萃取,无水MgSO4干燥有机相,减压旋干溶剂。得到的粗产物采用柱层析(PE/EA = 15/1 – 13/1 – 10/1)分离,得到淡黄色固体2 858 mg (Rf = 0.52, PE/EA = 5:1),产率79%。1H NMR (400 MHz, CDCl3): δ (ppm) 7.31 (m, 10H, -(C6H5)2), 4.57 (m, 2H), 4.50 (m, 2H), 4.41 (d, J = 18.0 Hz, 1H), 4.30 (m, 1H,), 4.26 (d, J = 18.2 Hz, 1H), 4.07 (dd, J = 6.1 Hz, J = 8.8 Hz, 1H,), 2.74 (m, 4H), 2.22 (m, 2H), 1.94 (m, 2H).13C NMR (100.6 MHz, CDCl3): δ (ppm) 208.3, 137.2, 128.6, 128.5, 128.2, 128.1, 128.0, 80.2, 73.4, 73.2, 72.9, 42.1, 37.3, 29.0, 28.7, 25.8. HRMS (ESI-TOF) m/z: [M + H]+Calcd for C22H27O3S2 403.1402; Found 403.1387.
化合物3b的合成。Ar保护,室温下将化合物2 192.3 mg (0.50 mmol, 1 eq)溶于3.7 mL THF,然后缓慢滴加CF3SiMe3 220 μL (1.49 mmol, 3 eq),于15℃反应18 h,TLC监测反应物全部转化成中间产物后,加入497 μL四丁基氟化铵溶液(1 M in THF, 1 eq),反应5 min,TLC监测中间产物全部转化成目标产物后,1 mL CH3OH萃灭反应,旋走THF和CH3OH,CH2Cl2萃取产物,无水MgSO4干燥有机相,减压旋干溶剂。得到的粗产物采用柱层析(PE/EA = 60/1 – 30/1 – 20/1 – 15/1)分离,得到淡黄色固体3a 31 mg (Rf = 0.32, PE/EA = 10/1),产率52%和3b 122 mg (Rf = 0.30, PE/EA = 10/1),产率52%。1H NMR (400 MHz, CDCl3): δ (ppm) 7.37 – 7.30 (m, 10H, -(C6H5)2), 4.85 (d, J = 11.1 Hz, 1H), 4.68 (d, J = 11.0 Hz, 1H), 4.55 (dd, J = 12.2 Hz, J = 14.0 Hz, 2H), 4.27 – 4.24 (m, 1H), 4.00 (dd, J = 3.5 Hz, J = 10.9 Hz, 1H), 3.80 (d, J = 10.0 Hz, 1H), 3.60 (d, J = 10.0 Hz, 1H), 3.48 (s, 1H), 2.83 – 2.71 (m, 3H), 2.64 – 2.57 (m, 1H), 2.12 – 2.03 (m, 2H), 1.93 – 1.81 (m, 2H). 13C NMR(100.6 MHz, CDCl3): δ (ppm) 138.2, 137.1, 128.6, 128.5, 128.1, 128.0, 127.9, 127.7, 125.4 (q, J = 287.8 Hz, -CF3), 76.96 (q, J = 25.5 Hz, -(CF3)C), 75.8, 75.5, 73.8, 67.8, 43.3, 35.8, 30.0, 29.2, 25.9. HRMS (ESI-TOF) m/z: [M + H]+Calcd for C23H28F3O3S2 473.1432. 3b: 1H NMR (400 MHz, CDCl3) δ (ppm) 7.32 (m, 10H, -(C6H5)2), 4.66 (dd, J = 11.3 Hz, J = 15.4 Hz, 2H), 4.56 (t, J = 8.3 Hz, 2H), 4.26 (dd, J = 3.1 Hz, J = 8.8 Hz, 1H), 3.99 (dd, J = 4.8 Hz, J = 10.0 Hz, 1H), 3.73 (dd, J = 10.3 Hz, J = 36.1 Hz, 2H), 3.52 (s, 1H), 2.72 (m, 4H), 2.23 (m, 1H), 2.02 (m, 2H), 1.84 (m, 1H). 13C NMR (100.6 MHz, CDCl3): δ (ppm) 138.1, 137.1, 128.6, 128.5, 128.3, 128.1, 128.0, 127.9, 127.8, 127.6, 126.1 (q, J = 287.2 Hz, -CF3), 76.5, 75.8 (q, J = 26.09 Hz, -(CF3)C), 74.6, 73.9, 67.3, 44.0, 36.8, 30.1, 29.6, 25.9. HRMS (ESI-TOF) m/z: [M + H]+Calcd for C23H28F3O3S2 473.1432; Found 473.1458.
化合物4的合成。配制6 mL 80%的CH3CN溶液,将NBS 980 mg (5.5 mmol, 6 eq)溶解,总共取5 mL CH3CN溶解化合物3b 431.7 mg (0.91 mmol, 1 eq)。Ar保护下于5℃将溶解的3b逐滴滴加到NBS中,反应10 min,TLC监测反应结束后,6 mL饱和Na2S2O3溶液萃灭反应,减压旋走CH3CN。加入CH2Cl2萃取,用饱和NaHCO3溶液洗涤有机相,最后用饱和NaCl溶液洗涤有机相,无水MgSO4干燥,减压旋干溶剂。将得到的粗产物与无水吡啶共旋两次,于室温下溶于10 mL无水吡啶中,将反应体系转移到0℃,向反应体系中逐滴滴加Ac2O 0.33 mL (3.45 mmol, 11 eq)。滴加完成后,室温下反应2 h,TLC监测反应结束后,旋走吡啶,加入4 mL饱和NaHCO3溶液萃灭反应,CH2Cl2萃取,无水MgSO4干燥有机相。减压旋干溶剂,得到的粗产物采用柱层析分离(PE/EA = 40/1 – 20/1 – 10/1),最后得到白色固体4 338 mg (Rf = 0.42, PE/EA = 10/1),产率87.4%。1H NMR (400 MHz, CDCl3): δ (ppm) 7.36 – 7.32 (m, 10H, -(C6H5)2), 6.36 (d, J = 5.2 Hz, 1H, H1′), 4.67 – 4.53 (m, 5H, -(CH2)2,H3′), 3.86 (d, J = 10.4 Hz, 1H, H5′), 3.73 (d, J = 10.4 Hz, 1H, H5′′), 2.53-2.46 (ddd, J = 5.4 Hz, J = 9.7 Hz, J = 15.1 Hz, 1H, H2′), 2.29 – 2.24 (m, 1H, H2′′), 2.02 (s, 3H, -CH3).13CNMR(100.6 MHz, CDCl3): δ (ppm) 169.8, 137.9, 137.4, 128.5, 128.4, 128.0, 127.6, 127.5, 124.5 (q, J = 284.3 Hz, -CF3), 96.5, 86.0 (q, J= 28.2 Hz, -(CF3)C), 77.7, 74.0, 73.3, 67.4, 38.1, 21.0. HRMS (ESI-TOF) m/z: C22H23F3NaO5, [M + Na]+ cal. 447.1395; found, 447.1380.
化合物5a和5b的合成。Ar保护下,取胸腺嘧啶2.6 g (20.8 mmol, 1.5 eq)加入到40 mL无水CH3CN中,于80℃油浴回流条件下,将N,O-双(三甲基硅基)乙酰胺10.7 mL (41.6 mmol, 3 eq)加入到反应体系中反应1 h,当白色固体完全溶解后,将化合物4 5.88 g (13.9 mmol, 1 eq)用20 mL无水CH3CN溶解后滴加到反应体系中,最后将TMSOTf 12.67 mL (69.3 mmol, 5 eq)滴加到反应体系中,于80℃回流反应2 h,TLC监测反应结束后,减压旋干CH3CN,将反应体系用饱和NaHCO3溶液萃灭,CH2Cl2萃取,饱和NaCl溶液洗涤有机相,无水MgSO4干燥。减压旋干溶剂,得到的粗产物采用柱层析分离(PE/EA = 2/1 – 1/1),得到目标产物5b 2.93 g (Rf = 0.31, PE/EA = 4/1),产率43%和另一个非对应异构体5a 2.86 g (Rf = 0.29, PE/ EA = 4/1),产率42%。5a: 1H NMR (400 MHz, CDCl3): δ (ppm) 9.33 – 8.98 (m, 1H, -NH), 7.35 – 7.25 (m, 10H, (-C6H5)2), 7.09 (s, 1H, H6), 6.46 (t, J = 6.6 Hz, 1H, H1′), 4.64 – 4.55 (m, 4H, (-CH2)2), 4.48 (dd, J = 4.3 Hz, J = 6.4 Hz, 1H, H3′), 3.91 (d, J = 10.6 Hz, 1H, H5′), 3.83 (d, J = 10.4 Hz, 1H, H5′′), 2.68 – 2.62 (m, 1H, H2′), 2.24 – 2.17 (m, 1H, H2′′), 1.92 (s, 3H, -CH3). 13C NMR (100.6 MHz, CDCl3) δ(ppm) 163.7, 150.3, 137.5, 136.9, 134.8, 128.6, 128.4, 128.2, 127.8, 127.7, 127.6, 124.5 (q, J = 285.4 Hz, -CF3), 111.8, 83.6 (q, J = 28.2 Hz, -(CF3)C), 85.8, 78.1, 74.0, 73.1, 67.6, 37.8, 12.7. HRMS (ESI-TOF) m/z:C25H26F3N2O5, [M + H]+cal. 491.1794; found, 491.1828. 5b 1H NMR (400 MHz, CDCl3): δ (ppm) 10.21 – 9.48 (m, 1H, -NH), 7.73 (s, 1H, H6), 7.43 – 7.36 (m, 10H, (-C6H5)2), 6.40 (dd, J = 6.2 Hz, J = 8.3 Hz, 1H, H1′), 4.74 – 4.58 (m, 4H, (-CH2)2), 4.51 (t, J = 7.7 Hz, 1H, H3′), 3.97 (t, J = 10.8 Hz, 2H, H5′, H5′′), 2.68 – 2.62 (m, 1H, H2′), 2.31 – 2.24 (m, 1H, H2′′), 1.55 (s, 3H, -CH3). 13C NMR (100.6 MHz, CDCl3) δ(ppm) 164.0, 150.7, 137.3, 137.0, 135.8, 128.7, 128.6, 128.3, 128.2, 127.9, 127.8, 124.6 (q, J = 285.2 Hz, -CF3),111.9, 85.2 (q, J = 27.5 Hz, -(CF3)C), 82.9, 77.6, 74.1, 73.5, 67.3,37.6, 12.1. HRMS (ESI-TOF) m/z:C25H26F3N2O5, [M + H]+cal. 491.1794; found, 491.1828.
化合物6的合成。H2保护下,取化合物5b 1.58 g (3.2 mmol, 1 eq)溶于20 mL无水CH3OH中,加入 PdCl2 2.1 g搅拌反应2 h,TLC监测反应结束后,使用硅藻土抽滤,除去不溶固体,减压旋干CH3OH。得到白色固体粗产物采用柱层析分离(PE/EA = 2/1-1/1-1/1.5-1/3),得到白色固体6 945 mg (Rf = 0.4, PE/ EA = 1/3),产率95%。1HNMR (400 MHz, D2O): δ (ppm) 7.32 (s, 1H, H6), 6.36 (t,J = 6.6 Hz, 1H, H1′), 4.88 (t,J = 6.2 Hz, 1H, H3′), 3.95 (d, J = 12.8 Hz, 1H, H5′), 3.84 (d, J = 12.8 Hz, 1H, H5′′), 2.58 – 2.44 (m, 2H, H2′, H2′′), 1.80 (s, 3H, -CH3). 13CNMR(100.6 MHz, D2O): δ (ppm) 166.2, 151.7, 136.8, 124.5 (q, J = 284.4 Hz, -CF3), 111.8, 86.4 (q, J= 26.5 Hz, -(CF3)C),85.3, 70.7, 59.3, 37.9, 11.5. 19FNMR (376 MHz, d6-DMSO): δ (ppm) -77.03.HRMS (ESI-TOF) m/z: C11H13F3N2NaO5, [M + Na]+ cal. 333.0674; found, 333.0661.
化合物7的合成。Ar气保护下,将化合物6 850 mg (2.74 mmol, 1 eq)溶于10 mL无水吡啶中,加入DMTr-Cl 928 mg (3.01 mmol, 1.1 eq),常温搅拌反应4 h,TLC监测反应结束后,加入3 mL甲醇萃灭反应,旋干体系,得到的粗产物采用柱层析分离(PE/EA = 2/1 – 1/1 – 1/1.5 – 1/3,加入1%三乙胺),最后得到白色固体7 1.39 g (Rf = 0.46, PE/EA = 1/1),产率83%。1H NMR (400 MHz,CDCl3) : δ (ppm) 9.51 (m, 1H, -NH), 7.43 – 7.31 (m, 9H, ArH), 7.14 (s, 1H, H6), 6.87 – 6.85 (m, 4H, ArH), 6.57 (dd, J = 6.5 Hz, J = 7.8 Hz, 1H, H1′), 4.78 (d, J = 5.7 Hz, 1H, H3′), 3.78 (s, 6H, (-OCH3)2), 3.68 (d, J = 9.9 Hz,1H, H5′), 3.43 (d, J = 9.8 Hz,1H, H5′′), 2.49 – 2.45 (m, 1H, H2′), 2.27 – 2.20 (m, 1H, H2′′), 1.92 (s, 3H,-CH3). 13C NMR (100.6 MHz, CDCl3) δ(ppm) 163.7, 158.9, 150.5, 143.6, 134.3, 129.8, 128.2, 127.7, 127.3, 124.8 (q, J = 284.4 Hz, -CF3), 113.6, 112.1, 87.8, 86.9 (q, J= 26.9 Hz, -(CF3)C), 85.5, 72.6, 55.2, 39.7, 12.8.HRMS (ESI-TOF) m/z:C32H31F3N2NaO7, [M + Na]+cal. 635.1981; found, 635.2054.
化合物8的合成。Ar气保护下,将化合物7 1.4 g (2.28 mmol, 1 eq)溶于15 mL无水CH2Cl2中,加入DIPEA 3 mL (18.28 mmol, 8 eq),常温搅拌3 min后,加入N,N-二异丙基氯代亚磷酰胺 2 mL (4.57 mmol, 4 eq),常温搅拌反应30 min。TLC监测反应结束后,加入2 mL甲醇萃灭反应,减压旋干体系。将得到的粗产物采用快速柱层析分离(PE/EA = 1/1,加入1%的三乙胺),得到粗产物1.58 g,将粗产物溶于2 mL CH2Cl2中,分别滴加到4个装有10 mL正己烷的样品瓶中沉淀,得到白色晶状固体8 1.38 g,产率75%。1H NMR (400 MHz,CDCl3): δ (ppm) 1H NMR (400 MHz,CDCl3): δ(ppm) 7.49 – 7.20 (m, 9H, ArH), 7.17 (s, 1H, H6), 6.86 – 6.84 (m, 4H, ArH), 6.52 (t, J = 6.6 Hz, 1H, H1′), 4.83 – 4.78 (m, 1H, H3′), 3.77 (s, 6H, (-OCH3)2), 3.53 – 3.47 (m, 5H), 3.09 (s, 1H), 2.68 – 2.62 (m, 1H, H2′), 2.54 – 2.52 (m, 1H, H2′′), 2.38 – 2.35 (m, 3H), 1.96 (s, 3H, -CH3), 1.06 – 1.04 (d, 12H, (-CH3)4). 31P NMR (162 MHz, CDCl3) δ: 150.781 (s, 1P). HRMS (ESI-TOF) m/z: C41H48F3N4NaO8P, [M + Na]+cal. 835.3060, found, 835.3058; C41H49F3N4O8P, [M + H]+ cal. 813.3240, found, 813.3243.
3. 结果与讨论
3.1. 反应路线的优化
本路线从已知的化合物1 [8] 出发,在对化合物1进行羟基氧化成羰基这一步,尝试了如图2所示的一些氧化反应。1) Swern氧化:反应利用二甲亚砜(DMSO)做氧化剂和有机碱(如三乙胺)在低温下与草酰氯协同作用将一级醇或二级醇氧化成醛或酮。由于Swern氧化反应条件严苛,得到的目标产物2的产率太低。2) 二碘酰基苯甲酸氧化:反应以乙腈为溶剂,反应温度80℃,回流的条件下反应2小时,原料消失,无目标产物出现,可能原因是反应条件剧烈,二碘酰基苯甲酸显酸性,使化合物1的结构不稳定而被破坏。3) Dess-Martin氧化:由于Dess-Martin氧化反应条件温和,在常温中就能进行。经过多次改进,发现当原料的投料为1 g左右,Dess-Martin为1.2 eq时,产率达到79%。
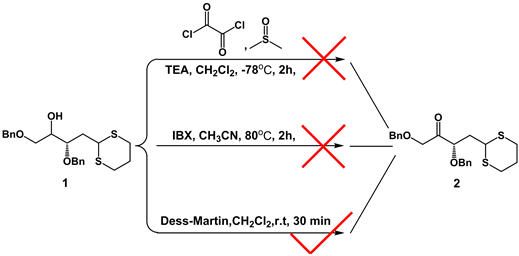
Figure 2. Optimization of the reaction route for the synthesis of compound 2
图2. 化合物2合成的反应路线优化
在将化合物1的C4′-羟基氧化成酮后,使用CF3SiMe3引入三氟甲基,然后用TBAF处理,得到一对非对映异构体3a和3b,为了提高反应的选择性,经过不断地尝试,最终发现当原料,CF3SiMe3和四丁基氟化铵当量比为1:3:1时,且反应温度控制恒温在15℃时,反应的选择性能达到4:1。其中主要的异构体3b经过两步化学反应得到一对非对映异构体5a和5b。在MeOH中将化合物5b在PdCl2/H2催化作用下脱除两个苄基保护基,得到C4'-CF3-α-L修饰的脱氧胸苷6,产率95%。最后通过对6的5′-OH使用DMTr保护和3′-OH与2-氰乙基N,N-二异丙基氯代亚磷酰胺反应,得到最终目标化合物8。
3.2. 非对映异构体5a,5b和化合物6的核磁数据分析
3.2.1. 非对映异构体5a,5b的核磁数据分析
通过1H谱,13C谱归属重要的H峰和C峰。然后通过2D NOESY实验(图3,图4),发现非对映异构体5b的H3′和H6有NOE效应,H3′和H5′没有NOE效应,说明H3′与H6空间距离相近,与H5′空间距离相远。H3′空间位置已知,位于糖环面上,所以碱基与H3′共面,位于糖环面上,H5′与H3′异面,位于糖环面下,证明了5b为目标化合物C4′-CF3-α-L脱氧胸苷。而5a的H3′与H1′有NOE效应,与H6和H5′没有NOE效应。说明H1′与H3′共面,位于糖环面上。H6和H5′与H3′异面,说明5a的碱基位于糖环面下,所以5a为C4′-CF3-β-L脱氧胸苷,是5b的非对映异构体。
3.2.2. 化合物6的核磁数据分析
为了进一步验证非对映异构体5b是需要的目标化合物,将5b的3′和5′的保护基团苄基在PdCl2/H2催化作用下脱除,得到化合物6,通过NOESY谱图(图5),发现H3′与H6有NOE效应,H3′与H5′和H1′没有NOE效应,证明了化合物6也是C4′-CF3-α-L脱氧胸苷。同时19F谱图显示,在−75到−80 ppm之间出现了F原子的特征峰(图6),也证明了化合物6是目标中间产物。
3.3. 目标化合物8的表征
首先通过ESI-TOF高分辨质谱对化合物8的相对分子质量进行表征(图7),化合物8加上H+离子和Na+离子的相对分子质量的理论值分别是813.3240和835.3060,实际测试得到的化合物8加上Na+离子相对分子质量是813.3243和835.3058,与理论值吻合。
然后通过对化合物8核磁谱解数据分析,发现化合物8的氢原子个数与氢谱中氢原子个数刚好是一致的。最后通过对化合物8进行31P谱的分析,发现在150.781 ppm处出现一个单峰(图8),与三价P原子的理论核磁出峰位置相一致,证明化合物8就是最终的目标化合物。

Figure 3. The NOESY spectrum of compound 5b
图3. 化合物5b的NOESY谱图
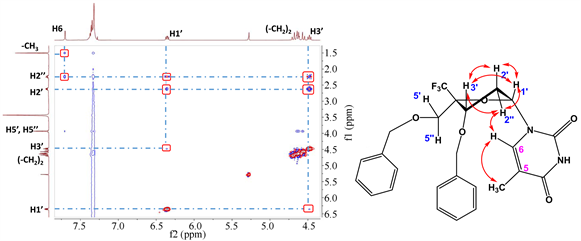
Figure 4. The NOESY spectrum of compound 5a
图4. 化合物5a的NOESY谱图

Figure 5. The NOESY spectrum of compound 6
图5. 化合物6的NOESY谱图

Figure 6. 19F NMR spectrum of compound 6
图6. 化合物6的19F NMR谱图

Figure 8. 31P NMR spectrum of compound 8
图8. 化合物8的31P NMR谱图
4. 结论
本文研究了以1,3-二(苄氧基)-4-(1,3-二噻烷-2-基)丁-2-酮为原料,通过7步反应得到了目标化合物8,该合成路线原料易得,通过优化条件,能使非对应异构体3b和3a具有4:1的反应选择性,且主要产物是所需要的目标中间产物。通过NOESY谱图对非对映异构体5a,5b和化合物6进行结构鉴定,能够确定5b和6为C4'-CF3-α-L-脱氧胸苷,最后通过1H NMR,31P NMR和ESI-MS高分辨液相质谱对8进行了结构鉴定,能确认化合物8是所需要的最终目标产物。
NOTES
*通讯作者。