1. 引言
S-(-)-α-苯乙胺是一种基础的碱性手性化合物,是制备磷霉素的中间体,并且是首选的酸性外消旋化合物拆分剂之一,尤其是近年来,手性工业快速发展,S-(-)-α-苯乙胺在洒石酸、苹果酸等工业拆分生产中得到了广泛的应用 [1] 。大部分S-(-)-α-苯乙胺由拆分外消旋体α-苯乙胺制得,但传统的合成拆分方法在合成产量和拆分后的光学纯度上都有所欠缺,因而对传统生产工艺的改进就变得更加重要。本文在传统合成拆分α-苯乙胺的基础上研究了反应物比例对合成外消旋α-苯乙胺的产量的影响以及拆分温度、拆分时间和搅拌速度对拆分后光学纯度的影响,提出最佳的工艺条件,并在最佳工艺条件下做出了最佳结果;并对产品的红外光谱和紫外光谱进行了分析。
2. 实验部分
2.1. 实验试剂
苯乙酮(AR,99%,天津有需化工有限公司);甲酸铵(AR,88%,长沙鑫本药业有限公司);氯仿(AR,99%,上海阳光试剂有限公司);盐酸(AR,36%~38%,南京化学试剂股份有限公司);氢氧化钠(AR,96%,南京化学试剂股份有限公司);甲苯(AR,99.5%,济南大晖化工科技有限公司);酒石酸(AR,99%,南京化学试剂股份有限公司);甲醇(AR,99.9%,山东德彦化工有限公司);无水硫酸钠(CP,98%,山东德彦化工有限公司);乙醚(AR,99.5%,安徽金邦医药化工有限公司)。
2.2. 外消旋α-苯乙胺的合成
在250 ml四口烧瓶中分别投加24 ml苯乙酮(99.9%)、40 g甲酸铵及十粒陶瓷拉西环,温度计插入液面下1 cm,在左侧口插入蛇形冷凝管改装成简单蒸馏装置,用油浴缓缓加热,温度升高后混合物缓慢熔成两层,温度升至150℃~155℃时混合物全部熔成液相。在沸腾状态下冷凝管下口不断馏出苯乙酮、水及碳酸铵,并且有气相氨气和二氧化碳气体放出。温度升到185℃,移除油浴加热装置,反应过程约需90 min。将馏出物移至球形分液漏斗进行分层处理,静置30 min后,分出下层液体(粗苯乙酮),并将分出的下层液体加入原反应瓶中。用油浴缓慢回报,控制温度182℃,反应90 min。
反应结束后,移除油浴回报装置,冷却,待温度降至室温,将反应液移入球形分液漏斗中,同时滴加30 ml蒸馏水反复洗涤,以除去水溶物。分出上层(粗品N-甲酰-α-苯乙胺)重新移入反应瓶中,分出的下层水相用氯仿萃取两次,每次用氯仿15 ml,并将两次萃取液一并移入反应瓶中。分别向反应瓶中滴加盐酸(37%),为防爆沸加十粒陶瓷拉西环,用油浴加热,馏出萃取剂,缓慢保持微回流50 min,使N-甲酰-α-苯乙胺完全水解。移除加热装置冷却,在温度降低过程中如有结晶析出,可以加入最少量的水使之溶解。再分别用15 ml氯仿萃取,并将3次萃取液合并,选取另外的500 ml的三口烧瓶回收氯仿,三口烧瓶余液移入500 ml烧瓶中。
将三口烧瓶移至冰浴中,当温度不再降低时,用恒压滴加漏斗缓慢滴加35%的碱液(先期配制好的氢氧化钠液体)并开启磁性搅拌器。约15 min滴加完成,并延迟反应30 min。反应结束后,移除冰浴,换成油浴加热,三口烧瓶加入十粒陶瓷拉西环,并加装蛇形冷凝管装置,收集液开始为碱性,收集至pH = 7为止。蒸馏结束后,收集液约为75 ml左右。
将上述蒸馏收集液移入250 ml梨形分流漏斗中,加入甲苯20 ml,进行萃取,共需萃取3次,并将3次萃取液合并后用氢氧化钠进行干燥,干燥后的萃取液移入250 ml三口烧瓶中加入十粒陶瓷拉西环,并加装蛇形冷凝管装置,先蒸馏出萃取剂,再改用空气冷凝管蒸馏,收集收集180℃~190℃馏分,产品总重约8 g~9 g,为无色透明油状液体。用橡塞塞密封,以备拆分实验使用。
2.3. 外消旋α-苯乙胺的拆分
对酒石酸进行溶解,在500 ml三口并烧瓶中,称取12.6 g (0.082 mol) (+)-酒石酸投加入烧瓶中,并量取200 ml的甲醇滴加入烧瓶中,使用油浴加热至微沸腾,开启电磁搅拌使其溶解。使用固体加料器,在搅拌状态下缓慢加入10 g α-苯乙胺。操作过程一定要缓慢小心,以免物料溢出造成损失,导致实验失败。反应结束后,冷却并用橡胶塞密封,静置一天以上,观察烧瓶内有白色棱状晶体析出。如发现析出物不是棱状晶体而是针状晶体,则应将针头晶体重新加热、溶解、冷却、结晶,处理至棱状结晶。然后将棱状结晶移至抽滤瓶中,加30 ml冷甲醇洗涤后抽滤,放入真空干燥箱中干燥,干燥后稳重得(-)-胺(+)-酒石酸盐约8 g。将本次实验的晶体约8 g,投入到250 ml的三口烧瓶中,滴加30 ml水,开启电磁搅拌,使其部分溶解,再使用恒压滴加漏斗,滴加50%氢氧化钠溶液,搅拌使晶体全部溶解为止。然后将溶解物转移至梨形分流漏斗中,分两次用乙醚进行萃取,乙醚用量每次20 ml,两次萃取液合并后用无水硫酸钠干燥。
将干燥后得醚萃取液用滴液漏斗分批转入25 ml圆底烧瓶中,在水浴中蒸去乙醚,然后蒸馏收集180℃~190℃馏分于一称重瓶中,产量约2 g~2.5 g,用塞子塞住锥形瓶准备测定比旋光度。
3. 结果与讨论
3.1. 通过改变苯乙酮与甲酸胺的投料摩尔比提高(±)-α-苯乙胺的产量
研究表明,投料比对产率会产生重要的影响 [2] ,结果如图1,可见,将投料比从1:2改变至1:5时,胺的产量迅速上升,但在比例升至1:6之后曲线趋于平直,这是由于在反应中甲酸胺分解为甲酸和氨气,氨与羰基发生亲和加成,接着脱水生成亚胺,而甲酸再还原亚胺生成胺。由于生成的氨气很多还来不及发生加成便逸出了,减少了亚胺的量,因而提高甲酸胺的用量可以提高产量;而当甲酸胺的用量增大到一定的数量时,由于受到苯乙酮用量的制约,产量不再明显上升。为了达到原料的充分利用,无需再提高比例,则达到最佳产量的投料比例是苯乙酮与甲酸胺的比例为1:5。
3.2. 通过改变拆分时间来提高S-(-)-α-苯乙胺的光学纯度
在进行拆分实验的过程中发现,加入反应物(±)-α-苯乙胺后的拆分时间对拆分后胺的比旋光度和光学纯度有很大的影响 [3] ,比旋光度及光学纯度的数据见图2。可以看出,当加入(±)-α-苯乙胺后的反应时间由10 min升至20 min时,拆分后的光学纯度明显上升,光学纯度由56%上升至60.2%,这是由于随着拆分时间的延长,拆分剂和被拆分的外消旋体可以充分接触,并使反应更彻底;但当反应时间延至25 min时,拆分后的光学纯度在60.2%~60.6%之间波动,几乎不变,这表示反应已经充分进行了,无需再延长时间,所以20 min时最佳的拆分时间。
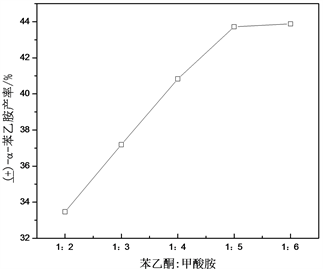
Figure 1. Effect of mole ratio of acetophenone to ammonium formate on the yield of α-phenylethylamine
图1. 苯乙酮与甲酸胺的投料摩尔比α-苯乙胺的产量的影响
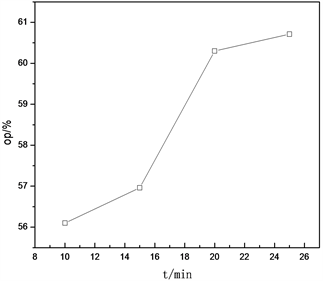
Figure 2. Effect of splitting time on optical purity of s-(-)-α-phenylethylamine
图2. 拆分时间对S-(-)-α-苯乙胺的光学纯度的影响
3.3. 通过改变拆分温度来提高S-(-)-α-苯乙胺的光学纯度 [4]
实验步骤中提到为使(+)-酒石酸溶解而加热至约
60 ℃
时,即混合物微沸腾时加入(±)-α-苯乙胺,但在实际操作中这个温度下很多酒石酸无法溶解,而且在拆分反应后放置一天后几乎没有任何结晶析出;考虑到甲醇的沸点是
64.5 ℃
,所以决定在
64.5 ℃
上下寻找合适的温度点进行这一条件实验,得到了显著的结果。数据及趋势见图3。可以发现,当拆分温度由
62 ℃
上升至
70 ℃
时,拆分后的光学纯度迅速上升,提高温度的效果十分显著,这是由于随着温度的升高,酒石酸得到了充分的溶解,从而使反应进行得更加充分,拆分的效果更加好;但是当温度升至
74 ℃
时,拆分效果迅速下降,且在实验过程中可以观察到,当加入(±)-α-苯乙胺后,三口烧瓶的内瓶壁上附着了一些白色的膏状物质,这可能是由于L-酒石酸在
74 ℃
受热而部分分解,减少了反应物,从而降低了拆分效果。由此看出,最佳的拆分温度是
70 ℃
。
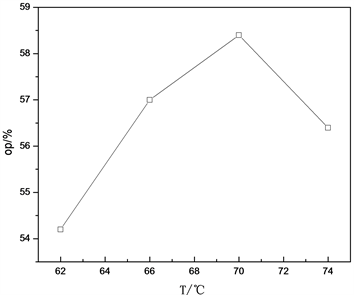
Figure 3. Effect of splitting temperature on optical purity of s-(-)-α-phenylethylamine
图3. 拆分温度对S-(-)-α-苯乙胺的光学纯度的影响
3.4. 通过改变拆分时的搅拌速度来提高S-(-)-α-苯乙胺的光学纯度 [5]
在进行前期的尝试性实验过程中,发现了在拆分过程中如果搅拌速度不一样的话,会引起从结晶形状到光学纯度等一系列结果的变化,所以决定考察搅拌速度对于拆分效果的影响。数据及趋势见图4,可以发现:当搅拌速度由185 r/min升到240 r/min时,拆分效果得到了明显的提高,这是由于搅拌是为了使酒石酸更快地溶解以及两种反应物充分的接触,而搅拌速度的加快促进了酒石酸的溶解和反应地进行;但是,可以看到,当搅拌速度上升到264 r/min时,拆分效果迅速下降,在实验中可以观察到,在这个条件下得到的晶体较少,大多数都呈粉末状,这是由于搅拌速度太高使得晶体过于细小,从而降低了拆分效果。由以上分析可知,拆分过程的最佳搅拌速度是240 r/min。
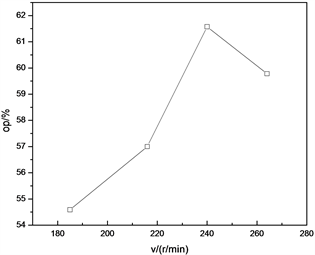
Figure 4. Effect of stirring speed on optical purity of S-(-)-α-phenylethylamine during splitting
图4. 拆分时的搅拌速度对S-(-)-α-苯乙胺的光学纯度的影响
3.5. 制备α-苯乙胺的红外光谱
由红外光谱图(图5)可以看出样品所含的官能团:① 3360和3300处有两个中等强度的吸收峰,是伯胺的N-H伸缩振动;② 3087,3062,3028这一组峰是苯环的C-H伸缩振动,且1650到1450的峰形尖锐的几个吸收峰是苯环的C=C伸缩振动(即骨架变形振动),这些就能够证明苯环的存在;729和696是面外弯曲振动,是单取代苯的证明。③ 1650-1590应是N-H弯曲振动的较强峰,但却被在这一区域有很强吸收的苯环的C=C伸缩振动而覆盖;1180处有一个强度较弱的吸收峰,这是C-N伸缩振动;1410处的另一个C-N伸缩振动也同样被苯环的C=C伸缩振动覆盖。通过(图6)样品的核磁图谱进行定性和定量分析各种有机物的成分、结构,与α-苯乙胺的核磁图(1HNMR)一致。
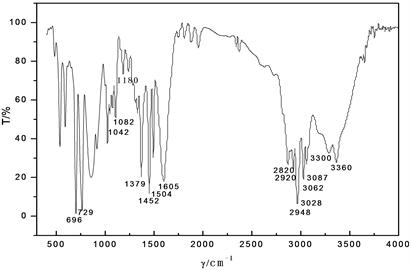
Figure 5. Infrared spectrogram of α-phenylethylamine
图5. α-苯乙胺的红外光谱图
3.6. 制备α-苯乙胺的核磁图(1HNMR)
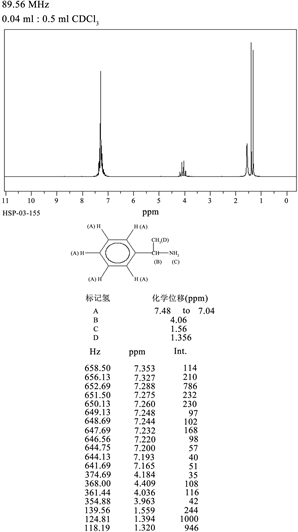
Figure 6. HNMR of α-phenylethylamine
图6. α-苯乙胺的核磁图(1HNMR)
4. 结论
由以上的条件实验确定了合成α-苯乙胺最佳苯乙酮与甲酸胺的投料摩尔比为1:5,拆分的最佳反应时间为20 min,反应最佳温度为
70 ℃
,拆分过程的最佳搅拌速度240 r/min。