1. 引言
最近几年,过渡金属催化C-H活化一直受到了人们的关注[1] [2] ,通过C-H活化我们可以在分子中直接形成C-C键得到许多有价值的化合物,反应原料不需要前活化,大大较少了成本,具有极高的原子经济性,这为一些复杂化合物的工业化生产奠定了基础。过渡金属催化C-H活化最为人们熟知的一个反应为Mizoroki-Heck反应 [3] ,反应使用芳基卤和烯烃反应,脱去一分子HX,实现芳基的烯烃化,该种反应为单方面C-H活化,原料芳基卤还是经过了前活化。为了真正实现C-H活化,人们开始转向双方面C-H活化,即原料都没有经过前活化,通过过渡金属催化以后脱去两个H原子实现C-C偶联,这样的反应称为脱氢Heck反应,这种反应涉及到两个C-H活化,难度较大,它是最近人们一直寻找的催化体系突破对象。
共轭二烯烃和和多稀类化合物存在于许多天然产物以及具有药物活性的化合物中,如维他命A,海鞘素、胡萝卜素、蚕蛾醇、绿色霉素等[4] [5] ,还存在于一些功能材料中,合成这些分子主要以下一些方法:羰基乙烯化反应叫做Wittig反应 [6] ,与此相类似的反应如Horner-Wadsworth-Emmons反应 [7] ,Julia稀化反应 [8] ,Peterson反应 [9] ,Stille偶联 [10] ,Heck稀基化 [11] ,Negishi偶联[12] 。最近,又有人报道了苯乙烯和丙烯酸酯的氧化偶联,生成共轭二烯烃,由此可以看到随着人们的研究深入,共轭二烯烃类化合物的合成越来越向直接C-H活化靠拢,但是还需要借助原料中的导向官能团辅助,如酯基、羰基,这些基团能在一定程度上能活化C-H键,又能起到C-H键的定位作用,因为在有机分子中存在大量的C-H键,许多C-H键的键能相似,所以在反应时存在反应选择性的问题,不可避免会形成同分异构体,对C-H键进行准确定位对合成特定的化合物来说很重要。
本文中,我们用苯乙烯为原料,用Pd(acac)2作催化剂,用廉价的Cu(OAc)2作氧化剂,添加AgF,并用低毒的乙酸乙酯作为溶剂,在100℃下脱氢偶联得到1,4-二苯基-1,3-丁二烯,分离产率为70%,并对苯乙烯做了三个底物的拓展,产率都在60%左右。我们并没有使用前活化的原料,也没有借助复杂的导向官能团辅助C-H活化,实现了脱氢Heck反应,为进一步改进苯乙烯脱氢偶联反应体系奠定了基础。
2. 实验部分
2.1. 主要仪器和试剂
实验试剂:乙酰丙酮钯、硫酸铜、氟化银、乙酸乙酯、苯乙烯以及其他实验相关的试剂。实验过程中所有的试剂都是从试剂公司购买直接购买,未经纯化直接使用。
主要仪器:1H NMR,13C NMR核磁数据采集采用Bruker Avance 300超导核磁共振仪,氘代溶剂为CDCl3,气相型号Shimadzu GC-2014系列,配有火焰离子化检测器,配有柱子的规格Rtx@-65 (30 m × 0.32 mm ID × 0.25 μm df),用联苯作内标,薄层色谱分析用的是硅胶板,Buchi旋转蒸发仪,SYNTHWARE压力管(10 ml)。产物通过柱层析分离,所使用的硅胶规格200~300目。
2.2. 反应条件的优化
苯乙烯脱氢偶联的反应式如图1所示。
如表1所示,通过苯乙烯脱氢偶联的条件优化可知:我们反应体系的最佳反应条件为1a (80 μl 0.7 mmol),Pd(acac)2 (7 mol%),Cu(OAc)2 (0.525 mmol 1.5 equiv),AgF (30 mol%),乙酸乙酯(2 ml),反应

Figure 1. Deyhdrogenative coupling of stryrene reaction equation
图1. 苯乙烯脱氢偶联反应式
表1. 条件优化
a气相产率,联苯作内标;b1a (0.7 mmol),Pd(acac)2 (5 mol%),Cu(OAc)2 (0.35 mmol 1 equiv),乙酸乙酯(2 ml)温度100℃,反应时间24 h;c1a (0.7 mmol),Pd(acac)2 (7 mol%),Cu(OAc)2 (0.525 mmol 1.5 equiv),温度100℃,反应时间24 h; d1a (0.7 mmol),Pd(acac)2 (7 mol%) Cu(OAc)2 (0.7 mmol 2 equiv)乙酸乙酯(2 ml)温度100℃,反应时间24 h;e体系中添加的添加剂的量均为20 mol%,除特别说明外。
温度为100℃,反应时间为24 h,体系用Ar保护,产物2a的气相产率最高为78%,经柱层析分离后的分离产率为70%,我们后来又尝试增加催化剂的量,氧化剂剂的量以及AgF的量,但是产率都在78%左右,并没有提高,所以就将最优的反应条件最终定为1a (0.7 mmol),Pd(acac)2 (7 mol%),Cu(OAc)2 (0.525 mmol 1.5 equiv),AgF (30 mol%),乙酸乙酯(2 ml)。降低反应温度到80℃,产率降温50%,当升高温度至130℃时副产物增多,产率为52%。反应时间增加到30 h时副产物增多,产率67%,减少反应时间到18 h,产率60%。
2.3. 实验步骤
取一个10 ml带有支管的高压反应管(SYNTHWARE),向试管中依次加入磁子、Pd(acac)2 (0.0075 g 7 mol%),Cu(OAc)2 (0.105 g,0.525 mmol,1.5 equiv)、AgF (0.013 g,30 mol%),盖上配套的黑色高压帽,不要拧紧,以备下一步通过支管向试管内部通Ar保护,打开与Ar伐相连的排气进气装置,将装置上的橡皮管与支管相连,先进行抽气,将管内的空气排净,10 min后将Ar伐打开,再将抽气伐关闭开进气阀,向试管内通Ar,30 s后关闭进气伐,再关闭Ar伐,打开抽气伐,如此循环2次,最后一次在通Ar时中将高压帽打开一个小口向管内快速注入86 μl的苯乙烯,再快速注入2 ml乙酸乙酯,最后快速将高压帽拧紧让高压管处于密封状态,关闭Ar伐及抽换气装置,将高压反应管放入100℃的油浴锅中,反应24 h。
反应完后反应液呈棕黄色澄清液,在试管底有少量的黑色固体,待反应液冷却后加入1 ml乙酸乙酯稀释,点板,展开剂为石油醚:乙酸乙酯(200:1),在紫外灯下展开图像,如图2所示。1为反应液展开结果,图中显示的是主要点,一些杂点未标出,2为苯乙烯展开结果,与1作对照。产物开始很淡,但是慢慢会显蓝色荧光,说明物质中含有双共轭结构。将反应液用细硅胶拌样,用柱层析分离,得到产物0.049 g,W = 70%,片状晶体。
2.4. 底物的拓展
我们对苯乙烯的偶联反应拓了三个底物,4-甲基苯乙烯、4-叔丁基苯乙烯和4-氯苯乙烯,在上述最优的反应条件下4-甲基苯乙烯反应后分离得到0.047 g (W = 58%),片状晶体。4-氯苯乙烯反应后分离得到0.071 g (W = 74%),白的固体。4-叔丁基苯乙烯反应后得到0.068 g (W = 61%),片状晶体。具体操作步骤同苯乙烯偶联反应,底物拓展如图3所示。我们也尝试了4-溴苯乙烯,反应后发现与该体系不兼容生成了其它产物。如何改进反应体系来扩大末端烯烃的偶联范围是我们下一步的研究方向。
3. 结果与讨论
通过大量的反应条件的筛选,我们得到了现在为止最优的反应条件,从所确定的反应条件来看,我们对该反应做了以下的猜想:Cu(OAc)2在反应过程中不仅起到了氧化剂的作用,而且Cu(OAc)2中的(OAc)−能起到配体的作用,稳定Pd(acac)2,AgF中的F−能在一定程度上活化C-H键,使C-H活化更容易进行,Ag+又能促进氧化过程的进行,在体系中起到辅助催化剂的作用。任何一个有机反应都是复杂的,以上只是猜想,下面给出一个我们猜想的反应机理图,如图4所示,有待进一步研究验证。
 R = methyl、Cl、t-butyl
R = methyl、Cl、t-butyl
Figure 3. Expansion of substrate
图3. 底物的拓展

Figure 4. Proposed catalytic mechanism
图4. 可能的反应机理图
核磁数据

1,4-二苯基-1,3-二烯:1H NMR (600 MHz, CDCl3) δ 7.44 (d, J = 7.3 Hz, 4H), 7.33 (t, J = 7.7 Hz, 4H), 7.23 (t, J = 7.3 Hz, 2H), 6.96 (dd, J = 11.9, 2.8 Hz, 2H), 6.67 (dd, J = 11.8, 2.8 Hz, 2H). 13C NMR (151 MHz, CDCl3) δ 135.43, 131.88, 131.26, 130.17, 129.01.
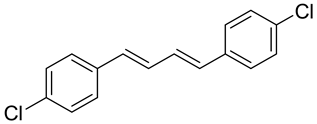
1,4-(4-氯苯基)-1,3-二烯:1H NMR (600 MHz, CDCl3) δ 7.34 (t, J = 6.9 Hz, 4H), 7.30 - 7.27 (m, 4H), 6.92 - 6.86 (m, 2H), 6.65 - 6.58 (m, 2H). 13C NMR (151 MHz, CDCl3) δ 138.35, 135.89, 134.58, 132.11, 131.49, 130.10.
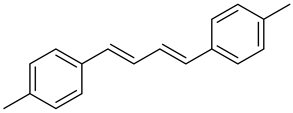
1,4-(4-甲基苯基)-1,3-丁二烯:1H NMR (600 MHz, CDCl3) δ 7.32 (d, J = 8.0 Hz, 4H), 7.13 (d, J = 8.0 Hz, 4H), 6.89 (dd, J = 11.9, 2.8 Hz, 2H), 6.61 (dd, J = 11.8, 2.8 Hz, 2H), 2.34 (s, 6H). 13C NMR (151 MHz, CDCl3) δ 139.99, 137.25, 134.88, 131.96, 131.13, 128.87, 23.49.

1,4-(4-叔丁基苯基)-1,3丁二烯:1H NMR (600 MHz, CDCl3) δ 7.37 (d, J = 8.5 Hz, 4H), 7.35 (d, J = 8.6 Hz, 4H), 6.91 (dd, J = 11.9, 2.8 Hz, 2H), 6.63 (dd, J = 11.9, 2.7 Hz, 2H), 1.32 (s, 18H). 13C NMR (151 MHz, CDCl3) δ 153.1, 137.32, 134.78, 131.39, 128.69, 128.21, 36.93, 33.91.