1. 引言
凡德他尼(Vandetanib, 1)化学名称N-(4-溴-2-氟苯基)-6-甲氧基-7-((1-甲基哌啶-4-基))喹唑啉-4-胺 [N-(4-bromo-2-fluorophenyl)-6-methoxy-7-((1-methylpiperidin-4-yl)methoxy)quinazolin-4-amine],是一种合成的苯胺喹唑啉类化合物,为口服的小分子络氨酸激酶抑制剂(TKI),可同时作用于肿瘤细胞表皮生长因子受体(EGFR)、血管内皮生长因子受体(VEGFR)和RET络氨酸激酶。表皮生长因子络氨酸激酶抑制剂(EGFR-TKI)不仅仅可抑制由EGF诱导的肿瘤细胞增殖,还可通过小调肿瘤细胞的血管生成因子以及抑制EGFR对肿瘤血管内皮细胞的信号传导,从而也可能具有抗血管生成作用 [1] - [3] 。美国FDA于2011年批准1上市,用于治疗成人晚期(转移型)不适合手术且疾病在持续发展的甲状腺髓样癌患者。国内还正在进行1治疗晚期NSCLC(非小细胞肺癌)的临床试验。
关于1的合成,根据其喹唑啉主环形成所用原料不同,主要有以下几条合成路线:Thomas等 [4] 以香草酸为起始原料,经苄基化反应成酯,然后硝化、还原,得到2-氨基-4-苄氧基-5-甲氧基苯甲酸苄酯,与醋酸甲脒环合,继而氯代、与4-溴-2-氟苯胺取代反应、脱苄基保护基等反应,总收率为3%;Hennequin等人 [5] [6] 采用价廉易得的香兰素为起始原料,经苄基保护、硝化、氧化、酰胺基化、硝基还原生成2-氨基-4-苄氧基-5-甲氧基苯甲酰胺,通过与氯化亚胺盐环合,继而氯代、与4-溴-2-氟苯胺反应、脱苄基等一系列反应,总收率仅为1.55%。以上路线除了收率较低,而且操作比较繁杂。本文在参考以上文献的基础上,采用改进的方法合成1,合成路线见Scheme 1。
合成路线中A链是以香兰素为起始原料,酚羟基的苄基保护生成3。传统的方法 [5] - [9] 3是先经过硝化然后再用高锰酸钾氧化醛基生成4,这样会出现高锰酸钾氧化产生大量的杂质而且后处理很麻烦,产率也太低(58%),原因可能是硝基位阻作用。本文将这两步颠倒过来,改变硝化反应条件,两步产率都在90%以上,而且产品较纯。对于6的成环的合成,有很多文献报道,以邻氨基苯甲酰胺、邻氨基苯甲酸酯的衍生物作为成环的原料,合成步骤较长,而且所用的环合试剂较贵,产率不高,本文直接在甲酰胺中,用无水甲酸作催化剂、脱水剂,于150℃反应4 h即可得到纯度较高且产率很高的产品。B链是以4-哌啶甲酸乙酯为原料,文献报道的合成路线都是先引入Boc基团对氨基进行保护,酯基还原然后脱Boc基团,再引入甲基。本文对此进行改进,首先对仲氨基进行甲基化生成12,这样既对氨基进行保护,而且又不用上、脱保护基,可以精简反应路线,提高产率,同时还避免了价格较贵的Boc酸酐和三氟乙酸的使用。然后通过硼氢化钠和氯化锌体系将12还原成醇13,方法适合工业化生产,避免了价格较贵的

Reagents and conditions: a) PhCH2Cl; b) KMnO4; c) HNO3; d) Fe, AcOH; e) HCONH2/HCOOH; f) SOCl2/DMF; g) ; h) TFA; TosCl
Scheme 1. Synthetic route of Vandetanib
Scheme 1. 凡德他尼的合成路线
四氢铝锂的使用。原料中的叔胺基结构还具有催化作用,其反应机理 [10] [11] 见图1。
2. 实验部分
熔点用毛细管法测定,温度计未经校正。核磁用Bruker ARX-300核磁共振仪测定;CARLOERBA -1106型元素分析仪;IR用NICOLET Impact 410型红外光谱仪,KBr压片;1H-NMR核磁共振用BRUKER
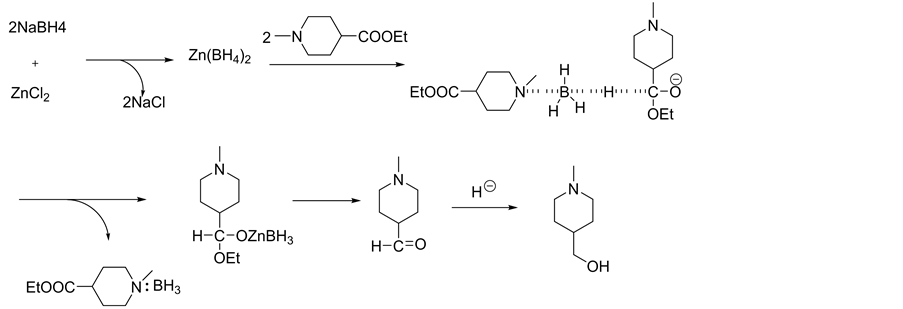
Figure 1. Reaction mechanism of compound 13
图1. 化合物13的还原机理
ARX300型核磁共振仪,CDCl3为溶剂,TMS为内标;质谱由岛津GC-MS 2050型气质联用仪(EI-MS)测定。
薄层层析(TLC)采用GF254薄层层析硅胶35 g (青岛海洋化工有限公司制造)与100 ml 0.8%的羧甲基纤维素钠(CMC-Na)混合均匀铺板,于110℃活化1 h后置于干燥器备用;柱层析采用硅胶为100~200目硅胶(青岛海洋化工有限公司制造)。TLC在ZF7型三用紫外分析仪(河南市巩义裕华仪器有限责任公司)下照射检测。所有试剂均为市售CP或AR产品,除特别说明外,均不经纯化处理直接使用。
2.1. 3-甲氧基-4-苄氧基苯甲醛(3)的制备
将香兰醛(15.2 g,0.1 mol)溶于乙醇(30 ml)中,加入氢氧化钾(6.1 g)和水(6 ml),加热搅拌至回流,然后用恒压漏斗滴加16 ml氯化苄,保持温度回流反应6 h,每隔一小时检测反应进程。趁热滤除无机盐,放入冰箱过夜,第二天抽滤析出的结晶,用乙醇洗涤,真空干燥得20.3 g黄色晶体3,收率84%,mp61~63℃ (文献 [12] :mp63~64℃)。
2.2. 3-甲氧基-4-苄氧基苯甲酸(4)的制备
将黄色晶体3 (14.7 g, 58 mmol),15 g KMnO4加入三口烧瓶中,并溶于170 ml丙酮和150 ml水的混合溶液中,加热至回流1 h,抽滤、减压蒸去丙酮,再抽滤除去不容物,滴加10% HCl溶液,调PH到2~3,有大量白色沉淀产生,抽滤、水洗、烘干得14.8 g白色粉末4,收率93.5%,mp170~172℃ (文献 [13] :mp171~173℃)。
2.3. 4-苄氧基-5-甲氧基-2-硝基苯甲酸(5)的制备
将化合物4 (14.8 g, 62 mmol)慢慢地加入到15℃ 150 ml浓硝酸中,保持温度反应1 h,得黄色乳浊液,倒入到200 ml冰水混合物中,有大量黄色沉淀生成,过滤收集沉淀,烘干后用乙醇重结晶得15.2 g纯品4,收率78.5%,mp161~163℃ (文献 [5] [6] 163℃~165℃)。1H-NMR (CDCl3) δ: 11 (s, 1 H, -COOH), 7.08 (s, 1 H, 6-H), 7.26 (s, 1 H, 3-H), 7.34~7.51 (m, 5 H, Ph-H), 5.2 (s, 2 H, Ph-CH2-O-), 3.9 (s, Ph-OCH3)。
2.4. 2-氨基-4-苄氧基-5-甲氧基苯甲酸(6)的制备
称量5 (5 g, 12.5 mmol)溶于150 ml冰醋酸中,搅拌升温至90℃时,慢慢于20 min内加入7 g Fe粉(已活化),反应3 h,抽滤,并用少量的冰醋酸洗涤滤饼,得棕色澄清溶液,加入150 ml冷水,有大量浅棕色沉淀生成,用150 ml CH2Cl2 (3 × 50 ml)萃取,合并有机层,真空旋干得粘稠液,然后加入10% HCl 50 ml,有少量沉淀生成,抽滤,滤液用15% NaOH溶液调pH = 12,有沉淀颗粒生成,置冰箱中冷却后抽滤即得浅棕色固体6,真空干燥后称重3.1 g,产率70.1%。 mp102~105℃ (文献 [7] 102℃)。1H-NMR (CDCl3) δ: 11 (s, 1 H, -COOH), 7.34~7.75 (m, 5 H, Ph-H), 5.22 (s, 2 H, Ph-CH2-O), 3.9 (s, 3 H, -OCH3), 3.4 (s, 2 H, -NH2)。
2.5. 7-苄氧基-6-甲氧基喹唑啉-4-酮(7)的制备
将化合物6 (0.8 g, 3 mmol)加入10 ml甲酰胺的三口烧瓶中,再加入1.5 ml无水甲酸,N2保护,于150℃下反应4 h后得澄清的橘黄色溶液,冷却到室温,将其倒入50 ml冰水混合物中,有大量浅棕色沉淀生成,用CH2Cl2 (3 × 10 ml)萃取,取有机层,用清水洗三次,再用无水硫酸钠干燥,减压蒸去溶剂并干燥得0.69 g浅棕色固体7,产率87.3%。1H-NMR (DMSO-d6) δ:7.91 (s, 1 H, 2-H), 7.23 (s, 1 H, 8-H), 7.47 (s, 1 H, 5-H), 7.35~7.44 (m, 5 H, Ph-H), 4.06 (s, 1 H, N-H), 5.26 (s, 2 H, Ph-CH2-O), 3.88 (s, 3 H, -OCH3)。
2.6. 7-苄氧基-4-氯-6-甲氧基喹唑啉(8)的制备
将化合物7 (1.8 g, 6.4 mmol)溶于20 ml重蒸的SOCl2溶液中,再滴加2滴DMF,得青黄色混浊液,加热至回流,缓慢滴加0.5 ml吡啶,回流至溶液澄清,大约1 h反应完全,冷却至室温,缓慢倒入100 ml冰水中,搅拌,析出黄色固体,抽滤并用清水洗涤滤饼,烘干得1.86 g,产率96.9%。1H-NMR (CDCl3) δ: 7.97 (s, 2 H, Ph-H), 7.33~7.47 (m, 5 H, -OCH2-Ph-H), 7.27 (s, 1 H, 2-H), 5.26 (s, 2 H, -CH2-), 3.88 (s, 3 H, -OCH3)。
2.7. 4-(4-溴-2-氟苯氨基)-7-苄氧基-6-甲氧基喹唑啉(9)的制备
4-溴-2-氟苯胺的制备:将四丁基溴化铵(3.22 g, 10 mmol)溶于10 ml氯仿中,在15 min内滴入含溴素(0.53 ml, 10 mmol)的氯仿(2.5 ml)溶液,滴加完毕室温搅拌30 min后慢慢滴入0.82 ml邻氟苯胺,温度控制在40℃以下,滴毕室温搅拌15 min后抽滤,得到白色粉末状的氢溴酸盐,再溶于水中得澄清透明溶液,滴加15% NaOH溶液调PH = 12,有白色固体析出,用乙酸乙酯(3 × 10 ml)萃取,收集有机层,无水硫酸钠干燥,减压蒸去溶剂得黄色油状液体,置于冰箱中析晶,得1.13 g,产率70.2%。mp40~42℃ (文献 [14] :mp41~42℃)。1H-NMR (CDCl3) δ: 7.14 (dd, J = 10.7, 2.1 Hz, 1 H), 7.05 (ddd, J = 9.1, 0.9, 0.4 Hz, 1 H), 6.66 (t, J = 8.9 Hz, 1 H), 3.60 (d, -NH2, 2 H)。
将化合物8 (0.95 g, 3.2 mmol), 4-溴-2-氟苯胺(0.7 g, 3.6 mmol),22 ml异丙醇加热至90℃,搅拌回流4 h,有沉淀生成,冷却至室温抽滤,并依次用异丙醇、THF、CH2Cl2洗涤,烘干得白黄色产品0.973 g,产率75.8%。1H-NMR (DMSO-d6) δ: 8.27 (s, 1 H, 2-H), 7.26~7.50 (m, 10 H, Ph-H), 5.27 (s, 2 H, Ph-CH2-O), 4.0 (s, 1 H, -NH-), 3.89 (s, 3 H, -OCH3)。
2.8. 4-(4-溴-2-氟苯氨基)-7-羟基-6-甲氧基喹唑啉(10)的制备
将化合物9 (0.973 g, 2.2 mmol),N2保护作用下在10 ml三氟乙酸中回流2 h,冷却至室温,缓慢倒入15 ml冰水中,有黄绿色沉淀生成,抽滤将滤饼溶于40 ml甲醇中,有少量不容,过滤取澄清滤液,用氨水调PH = 9,旋转蒸发,有青黄色沉淀生成,过滤,将滤饼真空干燥得0.425 g产品,产率60%。1H-NMR (DMSO-d6) δ: 8.29 (s, 1 H, 2-H), 7.07~7.78 (m, 5 H, Ph-H), 4.06 (s, 1 H, -NH-), 5.0 (s, 1 H, -OH), 3.95 (s, 3 H, -OCH3)。
2.9. N-甲基-4-哌啶甲醇(13)的制备
将4-哌啶甲酸乙酯(1 g, 6.4 mmol)慢慢滴加到0℃的甲酸溶液(5 ml)中,然后再滴加5 ml 37%甲醛溶液,维持在0℃搅拌15 min,升温至回流反应24 h后减压蒸除过量的甲醛和甲酸,残夜溶于10 ml CH2Cl2溶液中,加入0.5 g NaHCO3,有气泡生成,继续搅拌1 h后过滤,将滤液减压蒸馏得0.86 g浅黄色油状液体N-甲基-4-哌啶甲酸乙酯12,产率78.9%,不需要纯化直接投入下一步的反应。
将化合物12 (0.86 g, 5 mmol),0.3 g NaBH4,0.51 g ZnCl2投入到20 ml的无水THF中,回流2 h,反应液冷却至室温,缓慢滴加50 ml 10% NH4Cl溶液,有大量气泡生成,然后用氯仿(15 ml × 2)萃取,有机层依次用饱和食盐水、清水洗涤后经无水硫酸钠干燥,减压蒸干溶剂得0.45 g浅黄色油状液体13,产率69.2%。1H-NMR (CDCl3) δ: 3.66 (s, 1 H, -OH), 3.42~3.45 (d, J = 6, 2 H, -CH2-O), 2.84~2.87 (d, J = 9, 2 H, 2-CH2-N-), 2.25 (t, 3 H, -CH3), 1.88~1.96 (d, J = 2.4, 2 H, 2-CH2-N-), 1.71~1.76 (d, J = 15, 2 H, 3-CH2-), 1.24~1.33 (d, J = 2.7, 2 H, 3`-CH2-), 1.41~1.46 (s, 1 H, -CH-)。
2.10. 4-甲基苯磺酸-(N-甲基-4-哌啶)甲酯(14)的制备
将化合物13 (0.92 g, 7.2 mmol)溶于无水16 ml二氯甲烷中,加入3 ml无水三乙胺,冰浴冷却到0℃,分批加入对甲苯磺酰氯(7.2 mmol, 1.12 g),加完后在室温下搅拌4 h,用8 ml无水二氯甲烷稀释,分别用饱和的碳酸氢钠溶液洗、水洗后加无水硫酸钠干燥、减压蒸去溶剂,得1.55 g浅棕黄色油状液体14,产率77%。1H-NMR (CDCl3) δ: 7.26~7.29 (d, J = 9, 2 H, 2,2′-Ph-H), 7.67~7.79 (d, J = 3.6, 2 H, 3,3′-Ph-H), 3.21~3.23 (d, J = 6, 2 H, -O-CH2-), 2.41~2.47 (t, J = 12, 3 H, Ph-CH3), 1.71 (t, 3 H, N-H), 1.25 (s, 1 H, -CH-), 1.10~1.14 (m, 8 H, -CH2-)。
2.11. 4-(4-溴-2-氟苯胺基)-6-甲氧基-7-[(1-甲基哌啶-4-基)甲氧基]喹唑啉(凡德他尼,1)的制备
将化合物10 (2.0 g, 5.22 mmol)、化合物14 (1.7 g, 5.91 mmol)、K2CO3 (1.5 g)溶于20 ml DMF中,室温搅拌10 min后,氮气保护下升温至95℃反应2 h,然后用40 ml冰水稀释反应液,有米黄色沉淀生成,过滤,用乙醇两次重结晶得1.33 g米白色粉末状化合物1,产率47%,mp240~243℃。IR (KBr): 3174, 2932, 2837, 1618, 1578, 1503, 1456, 1422, 1389, 1365, 1242, 998, 777 cm−1。1H-NMR (DMSO-d6) δ: 8.39 (s, 1 H, 2-H), 7.87 (s, 1 H, 8-Ph-H), 7.67 (s, 1 H, 5-Ph-H), 7.49~7.64 (m, 3 H, 3′,5′,6′-Ph-H), 6.24 (s, 1 H, -NH-), 3.95 (m, 5 H, -OCH3, O-CH2-), 2.73 (m, 4 H, -CH3, -CH-), 1.98 (m, 4 H, 3′,5′-CH2-), 1.17 (m, 4 H, 2′,6′-CH2-)。HR-MS Calcd for C22H24BrFN4O2: 475.1139, Found: 475.3804。
3. 结论
A链中,重要中间体7的总产率为33.1%,比专利的产率[9] 高15.5%,合成步骤精简一步而且所需试剂价格便宜。B链的合成总产率35.1%,合成步骤省去了BOC酸酐的保护,反应步骤缩短,产率提高,操作简便适合工业化生产。