1. 引言
PI3K信号通路是癌细胞中常见的异常表达信号通路,其中PI3K,磷脂酰肌醇3-激酶,作为中间信号分子介导了PI3K/AKT/mTOR信号通路的发生,受多种致癌基因和生长因子受体的调控,现在普遍认为,PI3K信号通路的异常激活与人类癌症、免疫紊乱、神经紊乱等有密切关系。磷脂酰肌醇3-激酶蛋白家族参与多种细胞生理调节活动,如增殖、分化、代谢、迁移、分泌等 [1]。PI3K在具有磷脂酰肌醇激酶活性的同时,也具有丝氨酸和苏氨酸的活性。到目前为止,所发现的PI3K的亚型主要如(图1)所示,共分为三类,其中I类研究最为广泛,I类PI3Ks为异质二聚体,可细分为IA和IB两个类型,在结构上,由一个调节亚基p85和一个催化亚基p110共同组成,其中催化亚基p110还有四种亚型结构,分别为p110α、p110β、p110δ以及p110γ。根据文献报道,p110α分布于各组织器官,与癌症密切相关。p110β也分布于各组织器官,与血栓的形成有关。p110δ分布于髓系,与肺部气道炎症疾病有关。p110γ也分布于髓系,与肾小球、肾炎、类风湿性关节炎有关 [4]。II类PI3Ks存在三种类型,分别为PI3KC2α、PI3KC2β和PI3KC3γ。III类PI3Ks为异二聚体蛋白。PI3K信号通路的异常激活主要有三个途径,一是PI3K催化亚基的突变或扩增,二是脂质磷酸酶PTEN的失活,三是受体扩增或者突变 [2]。
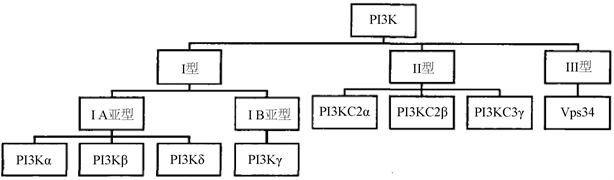
Figure 1. The classification of PI3K [3]
图1. PI3K的亚型分类 [3]
目前最为常见的PI3K抑制剂按照结构类型划分主要有十类,分别为甾类、苯并吡喃类、咪唑并喹啉类、喹喔啉类、喹啉类、噻吩并嘧啶类、咪唑并吡啶类、三嗪类、喹唑酮类以及其他类。
2. 甾类
2.1. Wortmannin (图2)
渥曼青霉素(Wortmannin)又名沃氏篮酶素或奥特曼宁,是一种在真菌中提取分离得到的天然产物,作为第一代PI3K抑制剂,在后期实验中,苏畅等 [5] 发现,由于Wortmannin对PI3K同工酶几乎没有选择性,所以它的毒性很大,并且Wortmannin的呋喃环在水中易开环水解而失活 [6]。基于Wortmannin的毒性大,水溶性差,结构不稳定等缺点,研究人员停止了对Wortmannin的临床开发。在分子模拟对接模型图中(图3),Wortmannin通过与ATP的C端和N端结合 [7],与参与磷酸盐结合反应的保守赖氨酸残基以共价键形式结合,是不可逆的PI3K抑制剂 [8]。Wormannin与PI3K活性位点是紧密结合的,既有疏水作用也有氢键作用,10位和13位上的甲基与其疏水口袋相连,这些疏水小囊在ATP结构中不被占据。Wortmannin与PI3K可以形成五种氢键的结合形式,主体甾环B环上的酮氧与PI3K-ATP结合位点的Asp-964的主链以及Ser-806的羟基形成作用力,B环上的羟基与Asp-964相互作用(该羟基是呋喃环上还未反应的醚氧),A环的酮氧与Ser-806的羟基相互作用,D环的酮氧与Val-882的主链相互作用。大量研究对Wortmannin进行了一系列结构改造,比如用羟基取代D环酮基会使得Val-882残基的主链氮原子与羟基上的氢形成附加的氢键;在16位引入溴、羟基、乙酰基等基团,会显著降低PI3K的亲和力;在11位上取代更多的亲脂基团,可以增加对PI3K的亲和力;当呋喃环被吡喃环取代时,与PI3K的亲和力下降 [7]。通过Wortmannin的结构改造可以发现,若想与PI3K紧密连接,可以选择性的引入亲脂性基团,
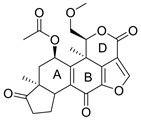
Figure 2. The chemical structure of Wortmannin
图2. 渥曼青霉素的化学结构式
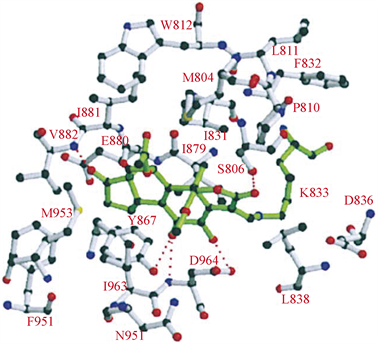
Figure 3. Predicted binding mode of compound Wormanninto PI3K [7]
图3. Wormannin与PI3K分子模拟对接模型图 [7]
避免亲水性基团的引入。若以不具有芳香性的吡喃环取代呋喃环,可能会干扰整个结构的共轭体系,降低抑制活性。
2.2. PX-866 (图4)
PX-866又名Sonolisib,是Filip Janku等人 [9] 根据Wortmannin结构研发的半合成I型PI3K抑制剂。目前I期临床的实验数据已被公布。PX-866是Wortmannin在仲胺的作用下,呋喃环开环而成的渥曼青霉素衍生物 [6],呋喃环的消失使其结构更加稳定 [3]。PX-866对PI3Kα、PI3Kβ、PI3Kδ、PI3Kγ四种亚型均有抑制活性,且相对于Wortmannin来说,PX-866降低了毒性,增大了抗肿瘤活性 [10],根据相关研究报道,PX-866的主要活性位置在20位碳原子上的二烯丙基取代基 [11]。
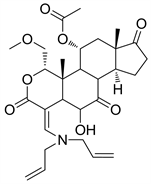
Figure 4. The chemical structure of PX-866
图4. PX-866的化学结构式
2.3. PWT-458 (图5)
PWT-458是Wortmannin中引入聚乙二醇侧链得到的聚乙二醇化的17-羟基麦芒素的衍生物,水溶性与稳定性均有提高 [6]。静脉注射给药后,聚乙二醇分子在体内被降解,释放出活性基团17-HWT (17-羟基麦芒素),达到抗肿瘤的活性 [8]。根据相关的研究报告,17-HWT比Wortmannin对人类PI3Kα的抑制活性更强,无论是单独使用还是作为联合用药使用,都具有良好的抗肿瘤活性 [12]。
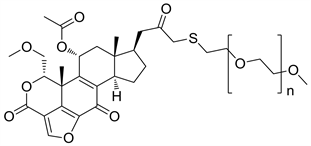
Figure 5. The chemical structure of PWT-458
图5. PWT-458的化学结构式
3. 苯并吡喃类
3.1. LY294002 (图6)
LY294002与Wortmannin同为第一代PI3K抑制剂,是以黄酮类槲皮素为结构基础设计的含有吗啉环取代的苯并吡喃类化合物,在体内对肿瘤诱导生长的血管和肿瘤生长具有明显的抑制作用 [13],但是由于LY294002溶解性差,半衰期短,以及相关的不良毒性,因此未能进入临床试验 [14]。在分子模拟对接模型图中(图7),LY294002的吗啉环与ATP酶复合物中的腺嘌呤部分重合,吗啉氧与残基Val-882的酰胺之间存在氢键。LY294002的苯并吡喃-4-酮与ATP腺嘌呤近似共平面 [7]。
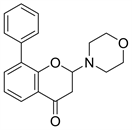
Figure 6. The chemical structure of LY294002
图6. LY294002的化学结构式
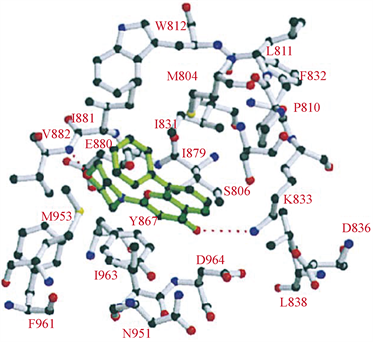
Figure 7. Predicted binding mode of compound LY294002 to PI3K [7]
图7. LY294002与PI3K分子模拟对接模型图 [7]
3.2. AZD8186 (图8)
AZD8186是阿斯利康公司研发的一种强效的PI3K选择性抑制剂,可以有效的抑制前列腺癌和三重阴性乳腺癌,AZD8186对PI3Kα的IC50值为35 nM,对PI3Kβ的IC50值为4 nM,对PI3Kδ的IC50值为12 nM,对PI3Kγ的IC50值为675 Nm [15]。因此AZD8186对PI3Kβ和PI3Kδ有较好的选择性。目前仍在临床I期。
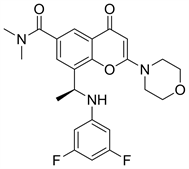
Figure 8. The chemical structure of AZD8186
图8. AZD8186的化学结构式
4. 咪唑并喹啉类
4.1. Copanlisib (库潘尼西) (图9)
Copanlisib又名BAY80-6946,是一种静脉注射的PI3K抑制剂,已经被证明了在各种细胞系和异种移植模型中具有强大的抗肿瘤功能以及促进细胞凋亡的活性 [9]。Copanlisib对PI3Kα和PI3Kδ都有可逆的抑制作用,Copanlisib会抑制Akt活化,增加肿瘤细胞凋亡。已批准上市。Copanlisib对骨髓瘤细胞有明显的凋亡诱导和生长抑制作用,也调节了骨髓瘤细胞周期减少骨髓瘤细胞的增殖 [16]。
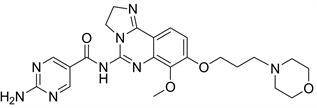
Figure 9. The chemical structure of Copanlisib
图9. Copanlisib的化学结构式
4.2. Dactolisib (图10)
Dactolisib又名NVP-BEZ235,Dactolisib是诺华制药研发的一种低分子量的咪唑并喹啉类化合物,通过与ATP结合位点竞争,可以有效的抑制PI3K的催化活性 [17]。现仍在临床II期。根据文献报道,作用机理是Dactolisib与该通路中的两种关键激酶PI3K和mTOR竟争性地结合ATP,进而影响PI3K/Akt/ mTOR信号通路下游相关分子的表达水平,抑制肿瘤的增殖 [18]。还有研究表明,Dactolisib能有效的阻断血管生成因子VERG诱导的血管内皮细胞增殖,因此也有效地抑制PI3K。还能抑制正常组织中的VERG的渗透性以及肿瘤组织中的血管渗漏 [19],降低肿瘤的IFP值。综上所述,Dactolisib有成为未来靶向PI3K抑制剂的潜力。
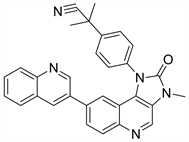
Figure 10. The chemical structure of Dactolisib
图10. Dactolisib的化学结构式
5. 喹喔啉类
5.1. Pilaralisib (图11)
Pilaralisib又名XL147或SAR245408,是一种有苯磺酰胺取代的喹喔啉类,主要作用于PI3Kα、PI3Kδ和PI3Kγ受体。目前仍在临床II期阶段。Pilaralisib主要用于细胞增殖并阻止AKT的磷酸化 [20]。
5.2. XL765 (图12)
对比XL765和XL147的化学结构,XL765结构中多了一个芳磺酰胺基,增强了与mTOR的竞争活性 [21]。现已进入临床II期阶段。
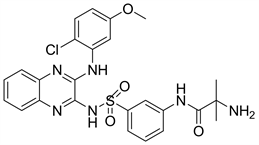
Figure 11. The chemical structure of Pilaralisib
图11. Pilaralisib的化学结构式
6. 喹啉类
6.1. NVP-BGT226 (图13)
有相关研究表明,在异种动物移植模型中,NVP-BGT226以剂量依赖性的方式显著延缓肿瘤生长。NVP-BGT226通过凋亡独立通路诱导癌细胞死亡,在体内和体外都有较强的抗头颈癌细胞的活性 [22]。
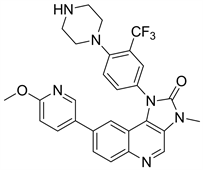
Figure 13. The chemical structure of NVP-BGT226
图13. NVP-BGT226的化学结构式
6.2. Omipalisib (图14)
Omipalisib又名GSK2126458或GSK458,是一种含有苯磺酰胺的喹啉类PI3K抑制剂,同时也对mTOR具有一定的亲和作用。根据分子模拟对接模型图(图15),Omipalisib与PI3K的结合中有几个作用点,在喹啉母核环上氮原子与PI3Kγ的Va1882以及PI3Kα的Val851 (图16)在氢键力的相互作用下连接。取代基吡啶环的氮原子通过一个水分子与PI3Kγ的Tyr867以及PI3Kα的Tyr836,PI3Kγ的Asp841以及PI3Kα的Asp810在氢键作用力下连接。磺酰胺基团上的氮原子与PI3Kγ的Lys833以及PI3Kα的Lys802在氢键的作用力下连接。根据以上的研究可以发现喹啉环、磺酰胺基、吡啶都与PI3Kγ以及PI3Kα形成氢键,为活性片段 [23]。有研究人员对此进行了结构改造,保留了喹啉母核以及磺酰胺基团,在母核喹啉环的4位引入亲水性基团或引入α,β-不饱和酰胺,在喹啉母核的3位引入亲水性侧链,并且替换了2,4-二氟苯基上的取代片段。经过虚拟分子与PI3Kα的分子模拟对接模型图显示,所改造的片段均与PI3Kα上的残基形成了新的氢键。且与mTOR进行了分子模拟对接模型图也形成了新的氢键。吕晓庆等人 [21] 据此设计了70个喹啉类的衍生物,经试验表明,大多衍生物对PI3Kα均有一定的抑制活性,甚至半数衍生物超过了Omipalisib对PI3Kα的抑制活性。目前Omipalisib已进入临床I期阶段。
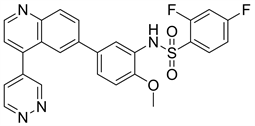
Figure 14. The chemical structure of Omipalisib
图14. Omipalisib的化学结构式

Figure 15. Predicted binding mode of compound Omipalisibto PI3Kγ [21]
图15. Omipalisib与PI3Kγ分子模拟对接模型图 [21]

Figure 16. Predicted binding mode of compound Omipalisibto PI3Kα [21]
图16. Omipalisib与PI3Kα分子模拟对接模型图 [21]
7. 噻吩并嘧啶类
7.1. Pictilisib (图17)
Pictilisib又名GDC-0941,Pictilisib作为一种单一药物或者于其他药物在联合用药过程中,已被证明对人类的胶质母细胞琳、小肠胃肠道间质瘤、乳腺癌等具有强大的抗肿瘤活性 [2]。Pictilisib还可以诱导人的肿瘤细胞株的凋亡,在携带p110α突变体的细胞中更为常见 [8]。Pictilisib与PI3Kγ结合的分子模拟对接结构图表明:母核噻吩并嘧啶的取代基吗啉环上的氧原子与Val-882骨架中的酰胺相互作用形成氢键,N端和C端之间的缝隙可以将母核噻吩并嘧啶结构放入。母核噻吩并嘧啶上的吲唑啉取代基插入到Ile-879和Asp-964所形成的口袋中,其中吲唑的两个氮原子与Tyr-867的苯酚基团之间也存在着作用力。母核噻吩并嘧啶环的6位上的取代基4-甲基磺酰基哌嗪甲基中的磺酰氨基上氧原子可以延伸到Lys-802 的侧链与Ala-805的酰胺基团的氮原子形成的氢键 [11]。对Pictilisib进行结构改造中,发现用2-氨基嘧啶替代吲哚基时,对PI3Kα的抑制活性得以保存,并且还发现对mTOR也有一定的抑制作用,之前Pictilisib对mTOR的IC50值为570 nM,用2-氨基密啶替代之后的化合物对mTOR的IC50值为29 nM [24]。
Yamanouchi与Pieamed [6] 的专利申请表明,噻吩并嘧啶类结构有较好的化学性质。
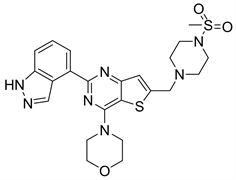
Figure 17. The chemical structure of Pictilisib
图17. Pictilisib的化学结构式
7.2. GDC0980 (图18)
其结构与Pictilisib很类似,用2-氨基嘧啶替代Pictilisib结构中的吲哚基团后,GDC0980具有PI3K-mTOR双重抑制活性 [25]。目前仍在临床II期。张睿等人 [26] 发现,在体外应用人早幼粒白血病细胞治疗白血病的实验中,将阿糖胞苷与GDC0980结合,再和阿糖胞苷联合用药,这种协同作用最强。
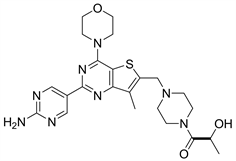
Figure 18. The chemical structure of GDC0980
图18. GDC0980的化学结构式
7.3. GNE477 (图19)
GNE477是在对Pictilisib的结构改造中发现的一种对PI3Kα和mTOR都有抑制作用的噻吩并嘧啶类化合物,GNE477对PI3Kα和mTOR的IC50值分别为2 nM和29 nM。GNE477是以2-氨基嘧啶取代Pictilisib的吲哚基团,研究人员发现其活性基团是有吗啉基取代的噻吩并嘧啶结构。化合物GNE477在其母环7位引入甲基可以提高口服生物利用度 [24]。
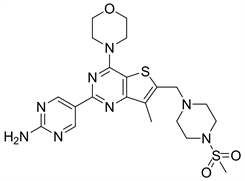
Figure 19. The chemical structure of GNE477
图19. GNE477的化学结构式
8. 咪唑并吡啶类
8.1. VS5584 (图20)
VS5584又名SB2343,是一种mTORC1/mTORC2双重选择性抑制剂,有研究表明,相比于非肿瘤干细胞,VS5584能够通过抑制PI3K/mTOR信号通路,更优先选择以体内干细胞为靶点来发挥抑制作用 [35]。目前仍在临床I期。
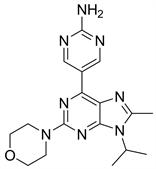
Figure 20. The chemical structure of VS5584
图20. VS5584的化学结构式
8.2. PKI402 (图21)
PKI402是惠氏公司研发的一种PI3K抑制剂,而且对mTOR也具有一定的抑制作用。具有抑制活性强,结构稳定等特点 [27]。
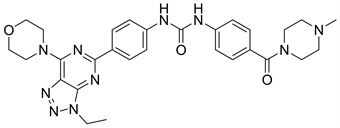
Figure 21. The chemical structure of PKI402
图21. PKI402的化学结构式
8.3. HS173 (图22)
HS173是一种咪唑并吡啶结构的选择性PI3Kα抑制剂,可以抑制PI3K信号转导通路,有明显的抗癌细胞增殖作用。HS173还会降低HIF-1α和VEGF的表达,这对于抑制肿瘤细胞诱导血管的生成起着重要的作用。因此HS173被认为是一种新型的抗癌药。在HS173与PI3Kα分子模拟对接模型图中(图23),HS173与残基Val-851、Tyr-863、Asp-810以及Lys-802以疏水键形式相互作用。HS173与PI3Kα之间的强亲和力是由于在ATP结合位点上同时建立了多个氢键和疏水键 [28]。

Figure 23. Predicted binding mode of compound HS173 to PI3Kα [28]
图23. HS173与PI3Kα分子模拟对接模型图 [28]
9. 三嗪类
9.1. ZSTK474 (图24)
ZSTK474为三嗪类PI3K抑制剂。正处于临床II期。有研究表明,ZSTJ474可以抑制A375细胞的增殖,抑制强度与浓度成正比 [29]。根据有关文献,ZSTK474与GDC-0941在JFCR39 (是Shingo Dan之前利用39个人类癌细胞系建立的信息型药物活性数据库)上表达了更多相似的指纹。因此ZSTK474与GDC-0941抑制谱高度相似 [14]。分子模拟对接模型图中,ZSTK-474与PI3K的活性位点形成3个氢键结合位点:三嗪结构2位的取代基苯并咪唑硝基与Val-882上的主链酰胺氮结合;三嗪结构6位的取代基吗啉环上的氧与Lys-802的侧链-NH结合;而另一侧吗啉环上的氧与Ser-806的-OH结合 [6]。
9.2. Gedatolisib (图25)
Gedatolisib又名PKI587或PF-05212384,是新型PI3K/mTOR抑制剂,对PI3Kα和PI3Kγ具有较高的抑制活性 [30]。现已进入临床III期。根据有关文献,吗啉环上的氧与PI3Kα催化域的Val-851以氢键形式结合,这也是吗啉氧致使化合物失去活性的主要原因之一,为代谢失活 [31]。
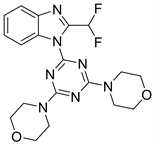
Figure 24. The chemical structure of ZSTK474
图24. ZSTK474的化学结构式
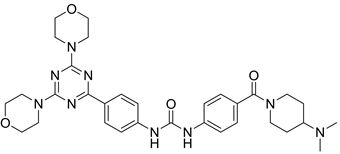
Figure 25. The chemical structure of Gedatolisib
图25. Gedatolisib的化学结构式
10. 喹唑酮类
10.1. Idelalisib (艾代拉里斯) (图26)
Idelalisib又名CAL-101或GS-1101,该药是FDA批准的首个选择性口服阻断磷脂酰肌醇-3激酶的抗癌药物,作用于PI3Kδ受体 [32] [33]。虽然Idlalisib对正常免疫细胞没有细胞毒性,但是改变了细胞因子的产生,这些细胞因子往往是单抗在体内初始给药后形成的可能会危及生命的细胞因子 [34]。重要的是,Idelalisib不会加速正常T/NK细胞的凋亡,也不会阻碍抗体依赖性细胞的细胞毒性。相关临床研究表明,Idelalisib与利妥昔单抗的联合应用或者是与苯达莫司汀的联合应用,这样联合用药的策略用来治疗慢性淋巴细胞白血病,似乎比利妥昔单抗与苯达莫司汀的联合用药效果要更好 [2]。
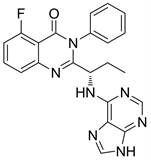
Figure 26. The chemical structure of Idelalisib
图26. Idelalisib的化学结构式
10.2. IPI145 (图27)
IPI145的化学结构与Idelalisib的化学结构很相似,在Idelalisib结构的基础上对氟原子和乙基进行了改造得到了IPI145。IPI145是p110δ选择性抑制剂,在较高的浓度下对p110γ也有一定的抑制作用。IPI145主要作用于人类T细胞,抑制其增殖 [30]。已在2018年批准上市。
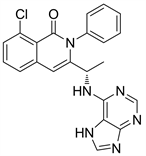
Figure 27. The chemical structure of IPI145
图27. IPI145的化学结构式
11. 其他类
除了上述罗列的几类之外,还有大环内酯类(Temsirolimus西罗莫司、Everolimus依维莫司)、丙酰胺类(Taselisib GD7C-0032)、苯并咪唑类(GSK2636771)、噻唑类(Alpelisib BYL719)、噻吩并吡啶类(GNE493)、二氢吡咯并嘧啶类(CH5132799)、咪唑并嘧啶类(PKI402)、吡唑啉并吡啶(IPI-549)、吡啶并嘧啶类(PF-04691502、AZD6482)等。
12. 前景与展望
通过上述PI3K信号通路抑制剂的结构以及与受体的结合位点,可以总结出一些经验,我们在进行结构改造的同时,可以考虑以下几个方面:1) 适当的引入亲脂性基团;2) 保持整个体系的共轭结构;3) 可以尝试利用聚乙二醇技术(能够增大稳定性、靶向性,延长半衰期,减少刺激性);4) 苯并吡喃-4-酮、吗啉环、噻吩并嘧啶都与ATP结构有部分的重合,可以作为PI3K抑制剂的一个潜在母核或取代片段;5) 噻吩并嘧啶结构与PI3Kγ的缝隙正好嵌合;6) 2-氨基嘧啶似乎是一个对mTOR具有一定抑制活性的取代基;7) 取代基与PI3K-ATP可以形成氢键的位点:① Asp-964与酮氧键及羟基的氧原子;② Ser-806与酮氧键及吗啉的氧原子;③ Val-882与酮氧键、羟基、吗啉、喹啉、硝基;④ Lys-833与吡啶的氮原子;⑤ Val-851与喹啉、吗啉;⑥ Tyr-836与吡啶的氮原子;⑦ Lys-802与磺酰胺、吗啉等。我们可以根据以上经验对PI3K信号通路抑制剂进行开发,增大活性,减小毒性,增加其靶向性。不难发现,随着PI3K信号通路抑制剂的发展,许多PI3K信号通路抑制剂已经进入了临床,但只有少数获批上市,比如说去年获批的库潘尼西,以及今年获批的Duvelisib,现在已经证实了PI3K信号通路一直积极是拥有巨大潜力的抗肿瘤药物,与此同时,PI3K/mTOR双靶点抑制剂也成为研究的热点。相信在不久的未来,单一药物治疗癌症会越来越普遍。
NOTES
*通讯作者。