摘要: 长QT综合征(long QT syndrome, LQTS)是一种常见的心律失常综合征,根据有无外部因素,可分为先天性和获得性,是以心电图QT间期延长,尖端扭转性室性心动过速,晕厥和猝死为主要临床特征的致命性心脏疾病。LQTS致死率高,早期识别及有效的治疗可减少恶性心律失常及猝死的发生。本文介绍1例多基因突变的LQTS,结合文献对其定义、诊断及治疗进行讨论。
Abstract:
Long QT syndrome (LQTS) is a common arrhythmia syndrome, which can be divided into congenital and acquired syndromes according to the presence of external factors. It is a fatal heart disease characterized by prolonged QT interval on electrocardiogram, tip torsion ventricular tachycardia, syncope and sudden death. LQTS has a high fatality rate, early recognition and effective treatment can reduce the incidence of malignant arrhythmia and sudden death. This paper reports a case of LQTS with multiple gene mutations, and discusses its definition, diagnosis and treatment based on the literature.
1. 引言
患者女,32岁,因“晕厥1次,胸闷、气促4次”于2022年7月8日入院,患者2022年7月7日12时于医院采核酸时情绪紧张,后突发晕厥,伴双眼上翻,无抽搐、口吐白沫、大小便失禁,予胸外按压1~2分钟后意识恢复,立即完善心电图提示QTc 531 ms (图1),后曾呕吐4次胃内容物,无呕血、喷射样呕吐,至当地县医院心内科住院,急查血钾2.2 mmol/l,未补钾,后患者共出现明显胸闷、气促4次,期间2次捕捉心电图提示尖端扭转性室速(图2),每次持续5~10秒,予补钾治疗后未在发作。患者为求进一步诊治,以“尖端扭转性室性心动过速”收住我科。病程中,患者精神、饮食、睡眠差,大小便正常。查体:BP 116/75 mmHg,心界不大,心率74次/分,未闻及杂音、额外心音及心包摩擦音。既往体健,否认糖尿病、高血压及心脏疾病史,否认特殊药物服用史,家族史无特殊。入院后急查BNP 696 pg/ml,cTnI 0.06 ng/ml,K+ 3.9 mmol/l,Ca2+ 1.18 mmol/l,Na+ 139 mmol/L,予补钾后K+维持在3.7~4.68 mmol/l,余电解质正常。多次复查心电图QTc在575 ms~631 ms。动态心电图:窦性心律不齐;窦房结至房室结内游走心律,最长QT间期683 ms,最大QTc 631 ms。24小时尿钾:11.2 mmol/l,血清醛固酮:卧位:4.2 ng/dl,立位32.5 ng/dl。血清肾素:卧位:13.63 uIu/ml,立位:166.6 uIu/ml。血清醛固酮、肾素比值正常。肾上腺CT:左侧肾上腺内侧支稍增粗。心脏彩超:射血分数和心脏结构未见异常。

Figure 1. ECG on July 7, 2022: Prolonged QT interval, altered S-T segment
图1. 2022年7月7日心电图:QT间期延长,S-T段改变

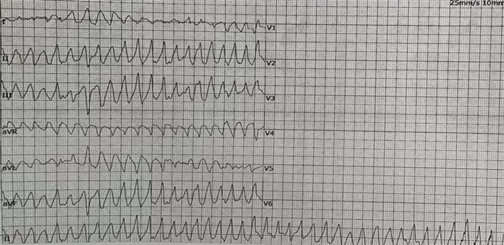
Figure 2. ECG on July 8, 2022: Ventricular tachycardia with prolonged QT interval
图2. 2022年7月8日心电图:室性心动过速,QT间期延长
2. 讨论
长QT综合征(long QT syndrome, LQTS)是一种常见的心律失常综合征,根据有无外部因素,可分为先天性和获得性,是以心电图QT间期延长,尖端扭转性室性心动过速,晕厥和猝死为主要临床特征的致命性心脏疾病,复极延长是LQTS的标志,又称为复极延迟综合征。1957年,LQTS首次在一个心脏结构正常的耳聋家庭中被发现,被描述为Jervell and Lange-Nielsen综合征 [1]。1963年和1964年,在没有耳聋的患者中也发现了类似的心电图,再次被描述为Romano-Ward综合征 [2] [3],而之后则使用了更通用的术语LQTS。60多年来,对LQTS的研究不断发展,逐步探索出了LQTS的分型、诊断及治疗方案。值得关注的是,虽然越来越多的LQTS患者得到了诊治,但仍有患者未能被早期识别、干预,而发生恶性心脏事件,所以,加强对LQTS的认识是必须的。
先天性长QT综合征(congenital long QT syndrome, cLQTS),是指先天携带某种基因突变,使心脏细胞离子通道发生变化,在没有心脏结构病变、各种药物及电解质紊乱等外部因素的下即出现QT间期延长,当紧张、情绪激动及声音刺激等因素出现时易发生室速、晕厥。在过去25年里,总报告17个LQTS遗传致病基因,其中包括ANK2、KCNE1、KCNE2在内的9个基因被归为证据有限或有争议的LQTS致病基因,只有3个最常见的亚型KCNQ1、KCNH2、SCN5A等被选为典型LQTS的遗传变异 [4]。在LQTS中有75%的病例具有致病变异,KCNQ1、KCNH2、SCN5A等3个“主要”基因占阳性突变病例的90% [5],已为其描述了特定的基因型–表型关系,分别是KCNQ1 (LQTS1)、KCNH2 (LQTS2)和SCN5A (LQTS3)。
获得性长QT综合征(acquired long QT syndrome, aLQTS),是由后天临床因素所引起,见于结构性心脏病、严重心动过缓、延长QT间期的药物、代谢性疾病(如低钾、低镁等)及其他疾病,其中低钾、药物影响较常见,触发离子通道使电流发生变化,发生恶性心律失常。近年来随着对LQTS研究深入,发现部分aLQTS也具有遗传病变,Hidekiltoh等人的研究表明,1/3的aLQTS患者携带cLQTS突变,KCNH2上的突变更常见 [6]。另外,在一项国际多中心LQTS临床基因再评估工作中,发现KCNE1和KCNE2不仅被报告为cLQTS的原因,还有许多报告支持它们在药物或电解质引起的LQTS病因学中的作用 [4],这些变异使aLQTS患者在其他QT延长因素存在下使携带者更易发生QT延长和扭转。
LQTS的诊断传统上依赖于QT间期延长及Schwartz评分(表1) [7] [8]。患者出现临床症状后,需要对临床症状、家族史、用药史、心电图、检验资料、影像学检查进行评估并确定继发性因素可能,同时确定QTc,进行Schwartz评分,若诊断LQTS可能性大,建议进一步行基因检测。
长QT综合征(LQTS)的诊断标准Schwartz评分:分数 ≥ 3.5分,LQTS为高概率;<3.5分和>1分,LQTS为中概率;≤1分,LQTS为低概率。
该患者于情绪紧张时晕厥1次,当时QTc 531 ms,低钾时出现尖端扭转性室速,Schwartz评分7分,明确诊断为LQTS。患者本次晕厥后呕吐数次,后查血钾低,既往无低钾及四肢瘫软病史,入院后完善24尿钾正常,经补钾后血钾维持正常;患者血压正常,立位肾素偏高,醛固酮水平及两者比值正常,结合肾上腺CT,诊断肾性低钾血症依据不足,出院后电话随访,患者多次复查血钾正常,最终考虑为胃肠丢失大量钾离子。患者送至当地医院时未及时纠正低血钾,考虑由低钾触发室速。入我院及时纠正血钾,虽未发室速,但QT间期仍未见明显缩短,最长QTc达631 ms,且结合查体、检验及心脏彩超已排除心脏结构病变、药物等继发因素,不排外存在遗传因素可能,进一步完善基因检测发现KCNQ1、ANK2、MYBPC3、LAMA4基因阳性。其中,KCNQ1 (LQTS1)为LQTS典型的致病基因,其突变致其编码的钾离子通道功能减弱或丧失,影响电流的变化使QT间期延长 [9],并且更易在运动、游泳及情绪激动等交感神经兴奋时出现室性心律失常 [10] [11]。该患者晕厥前情绪紧张,推测当时可能存在基因诱发交感神经兴奋因素,进一步诱发室性心动过速,导致晕厥。遗憾的是,未捕捉到晕厥发生时的心电图以进一步明确。ANK2突变可导致长QT综合征4型(LQTS4),属于引起LQTS中证据不足或存在争议的基因,而MYBPC3、LAMA4不是LQTS常见致病变异。
研究发现,MYBPC3主要和肥厚型心肌病(hypertrophic cardiomyopathy, HCM)相关,其编码的心脏肌球蛋白结合蛋白C (MyBP-C)发生突变是家族性HCM最常见的原因 [12]。LAMA4是编码层粘连蛋白α4的编码基因,参与多种肿瘤的发病,也是扩张性心肌病(DCM)及致心律失常性右心室心肌病/异型增生(ARVC/D)可能致病基因 [13] [14]。值得一提的是,MYBPC3、LAMA4均和心肌病相关,而国内未曾报道过LQTS中存在LAMA4突变,对于MYBPC3,国内也曾只报告过1例LQTS存在该突变 [15],两者现均尚未被纳入LQTS致病变异范围,而现于LQTS中发现LAMA4基因阳性,并再次出现MYBPC3突变,这2个基因是否为LQTS新的遗传致病变异是需要进一步研究的问题,这需要更大的临床样本。随着研究进展,希望MYBPC3、LAMA4基因能进一步丰富LQTS的遗传内容。
综合上述结果,KCNQ1最可能为该患者的致病基因,其次为ANK2,而其余2个基因是否参与该患者的发病仍不确定。患者明确诊断为LQTS,在无获得性因素时出现QT间期延长,基因检测阳性,考虑为先天性LQTS1型(合并LQTS4型可能),低钾、交感神经兴奋为其高危因素。
LQTS的治疗主要是β受体阻滞剂、左心交感切除术(left cardiac sympathetic denervation surgery, LCSD)以及植入心律转复除颤仪(implantable cardioverter defibrillator, ICD)。β受体阻滞剂是LQTS治疗的基石,能降低不良心脏事件的发生。该患者低钾触发室速,在服用β受体阻滞剂的同时,服用电解质补充剂,使血钾维持在正常偏高值,但应注意电解质过高的情况,并予尼可地尔缩短QT间期。若患者服药时仍有心律失常复发,还可考虑LCSD及ICD。LCSD是β受体阻滞剂的下一级治疗,可降低其心律失常发作的风险。植入ICD是LQTS的最终水平治疗,对于β受体阻滞剂和/或LCSD治疗效果不佳、有心脏骤停病史或高危患者可考虑ICD植入 [5] [16]。该患者晕厥、反复室速及QTc ≥ 500 ms及携带致病基因,属于高危患者,建议进一步植入ICD。另外,该患者应保持健康的生活习惯,避免紧张情绪及其他交感神经兴奋情况,预防LQTS继发性因素发生,特别是低钾。最后,该患者建议完善心脏MRI,动态复查心电图、心脏彩超及电解质等,继续寻找其他可能的室速、晕厥及低钾病因,若有不适,及时就诊。现患者已出院3月余,对患者进行电话随访,目前患者规律口服上述药物,未再出现晕厥、室速及其余不适。
3. 结论
对于LQTS的病人应该积极寻找一切病因,尤其是年轻患者,无论有无继发危险因素,强烈建议进一步完善基因检测,寻找遗传因素。不同类型的LQTS患者出现尖端扭转性室速的诱因不同,明确病因有利于个体化治疗。
NOTES
*通讯作者。