1. 引言
金属氧化物半导体由于其优异的化学稳定性、较高的丰富度和低廉的成本,已被广泛地应用于太阳能光催化制氢 [1] [2] [3] 。然而,大多数金属氧化物由于其较宽的能带结构,并不利于光催化制氢。这是因为较宽的能带结构限制了催化剂对可见光的吸收,而仅能吸收的紫外光谱区域的能量只占所有太阳光能量的3%~5%,其它绝大多数光能都无法参与光催化反应 [4] 。为了提高金属氧化物半导体对可见光的吸收,提高其光催化反应活性,在金属氧化物上负载贵金属,使其具有局域表面等离激元效应(LSPR),是提高光催化反应效率的有效方法之一 [5] [6] [7] [8] 。
最近,Ho [9] 等报道了负载Pd的超薄D-HNb3O8纳米片材料展现出了优异的光催化析氢活性。Ag作为一种常见的贵金属,同样具备LSPR效应 [10] 。受此启发,本文采用水热法制备了HNb3O8纳米片,然后通过光沉积和浸渍的方法在HNb3O8纳米片负载了一定含量的Ag。负载Ag的HNb3O8纳米片的能带结构和LSPR可以通过改变负载Ag的方式和改变Ag的比例来调节。此外,本文还研究了负载Ag的HNb3O8纳米片在不同波长范围的光照射区域下的光催化活性,实验结果显示,可见–近红外光照射提高了其光催化效率,这是由于光热效应促进了紫外光诱导的光催化活性。
2. 实验部分
2.1. 试剂和仪器
五氯化铌(NbCl5)、四甲基氢氧化铵(25%水溶液)、硝酸银、丙酮、无水乙醇和甲醇均为AR分析纯,实验过程中均直接使用,无需纯化处理。
本实验所用仪器如表1所示。
2.2. 催化剂制备及活性测试
HNb3O8纳米片的合成如示意图1所示:
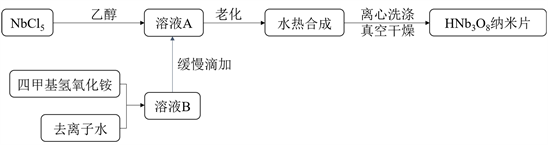
Figure 1. Schematic illustration of HNb3O8 nanosheets synthesis
图1. HNb3O8纳米片合成示意图
Ag/HNb3O8-pp纳米片光催化剂的合成如示意图2所示:
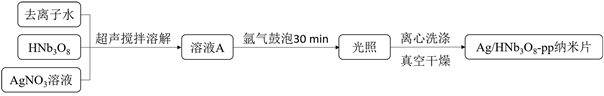
Figure 2. Schematic illustration of Ag/HNb3O8-pp nanosheets photocatalyst synthesis
图2. Ag/HNb3O8-pp纳米片光催化剂合成示意图
Ag/HNb3O8-DA纳米片光催化剂的合成如示意图3所示:
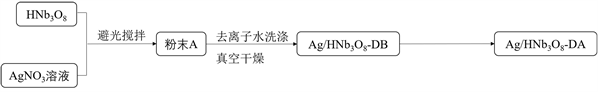
Figure 3. Schematic illustration of Ag/HNb3O8-DA nanosheets photocatalyst synthesis
图3. Ag/HNb3O8-DA纳米片光催化剂合成示意图
光催化活性测试:将20 mg催化剂分散在体积分数为20%的甲醇水溶液中,超声10分钟使其高分散,将石英管密封完好,氩气鼓泡30 min,确保反应体系内处于厌氧环境。用平行光照射石英管,用注射器抽取0.4 mL气体。最后,以氩气为载气,用配备热导检测器(TCD)的气相色谱仪,每隔一小时检测氢气的量。
3. 实验结果与讨论
3.1. 催化剂表征
图4是纯HNb3O8纳米片和负载Ag的HNb3O8纳米片催化剂的XRD图谱。纯HNb3O8纳米片在2θ角为7.30、14.14、17.60、23.22、25.54、27.06、30.60、35.76和46.70˚显示出了特征衍射峰,说明所合成的材料具有良好的结晶度,并且与Chen报道的HNb3O8纳米片的XRD特征衍射峰一致 [11] 。HNb3O8纳米片的主要衍射峰类似于层状H3ONb3O8 (JCPDS NO.44-0672)和块状HNb3O8 (JCPDS NO.37-0833)的衍射峰。与H3ONb3O8相比,我们所合成材料的(020)晶面所对应的衍射峰从2θ = 7.87˚偏移到了2θ = 7.30˚处。由布拉格方程可知,所合成的材料比块状HNb3O8具有更大的层间距,说明我们合成的材料具有片状结构。负载Ag后,可以观察到衍射峰的强度都有所降低,这表明结晶度有所下降。采用光沉积法制备的不同Ag负载量的Ag/HNb3O8-pp纳米片与纯HNb3O8纳米片的XRD衍射峰位置一致,没有检测到新的衍射峰,这是Ag负载量过少引起的。浸渍得到的Ag-HNb3O8反应前后的XRD图谱并不相同,Ag-HNb3O8-DB并没有检测到新的衍射峰,Ag-HNb3O8-DA在2θ = 37.33˚检测到新的衍射峰,且该衍射峰的位置对应Ag(0)的(111)晶面,这表明Ag-HNb3O8-DB经过一次反应后,催化剂中的Ag(I)被还原成Ag(0),Ag(0)是在催化反应过程中形成的。
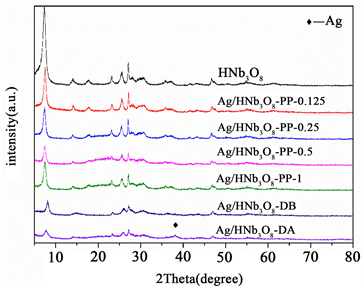
Figure 4. XRD patterns of various samples
图4. 不同样品的XRD图谱
用傅里叶变换红外技术(FT-IR)对HNb3O8纳米片和负载Ag的HNb3O8纳米片进行了表征,以获得催化剂的结构信息。如图5所示,在400~1000 cm−1范围内的红外吸收峰对应Nb3O8的骨架振动。在948、917、881 cm−1处的红外吸收峰归因于HNb3O8中Nb = O键的伸缩振动 [12] [13] 。680、634、566、529 cm−1的红外吸收峰归属于共角的NbO6八面体的伸缩振动。469 cm−1处的红外吸收峰对应于NbO6的弯曲振动峰;421 cm−1处的弱吸收峰则属于NbO6的振动模式 [12] 。无论是光沉积方法还是浸渍方法制备的样品,其红外吸收峰都没有发生改变。
Figure 5. FT-IR spectra of various samples
图5. 不同样品的红外光谱图谱
为了观察制备样品的形貌特征,对纯HNb3O8纳米片和负载Ag的HNb3O8纳米片样品进行了扫描电镜表征,结果如图6所示。从图中可以看出,我们合成的各种材料均由交错的二维纳米片组成,呈现出直径为1~4 μm的微米花形态。并且,无论是采用光沉积方法还是浸渍方法制备的催化剂,在负载Ag前后催化剂的形貌基本没有变化。从Ag/HNb3O8-DA的元素扫描图可以看出:Ag、Nb和O元素均匀分布在整个微花中。

Figure 6. SEM images of HNb3O8 (a); Ag/HNb3O8-pp-1 (b) and Ag/HNb3O8-DA (c, d); EDS elemental mapping of Ag/HNb3O8-DA (e, f, g)
图6. 样品HNb3O8 (a);Ag/HNb3O8-pp-1 (b);Ag/HNb3O8-DA (c, d)的SEM图;Ag/HNb3O8-DA的EDS图(e, f, g)
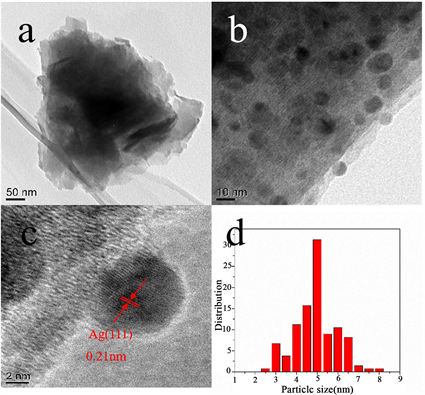
Figure 7. TEM (a, b) and HRTEM (c) images of Ag/HNb3O8-DA and (d) particle size distribution histogram of Ag/HNb3O8-DA
图7. Ag/HNb3O8-DA的透射电镜图(a, b);Ag/HNb3O8-DA的高分辨透射电镜图(c),(d) Ag的粒径分布图
为了进一步研究催化剂的形貌特征和微观结构,对Ag/HNb3O8-DA催化剂进行了透射电镜表征(图7)。催化剂的TEM图像(图7(a))显示Ag/HNb3O8-DA的形貌是一层层的纳米片,这与SEM表征结果一致。从图7(b)可以看出,一些小颗粒分布在一层层的HNb3O8纳米片上。其高分辨投射电镜表明这些颗粒的晶面间距为0.21 nm (图7(c)),对应Ag(0)的(111)晶面,由此得出这些颗粒是负载在HNb3O8纳米片上的Ag(0),XRD的表征结果也证明了同样的结果。对Ag(0)的粒径大小进行了统计,图7(d)是Ag(0)的粒径分布图。统计结果显示,Ag(0)的粒径大小在2.5~8 nm范围内,绝大多数Ag(0)的粒径为5 nm。
为了研究催化剂对光的吸收能力,对HNb3O8、Ag/HNb3O8-DB、Ag/HNb3O8-DA和Ag/HNb3O8-PP-1样品进行了紫外–可见近红外吸收光谱测试(UV-vis-NIR)。如图8(a)所示,纯HNb3O8纳米片在可见光区和近红外光区几乎没有吸收。Ag/HNb3O8-PP-1催化剂在可见光区有明显的吸收峰,且在近红外光区的吸收能力也有所增强。Ag/HNb3O8-DB与Ag/HNb3O8-DA有明显区别,未参与光催化析氢测试的催化剂Ag/HNb3O8-DB在可见光区和近红外光区几乎没有吸收,与纯HNb3O8纳米片对光的吸收能力基本一致;而经过析氢测试的催化剂Ag/HNb3O8-DA在可见光区检测到明显的吸收峰,近红外光区吸收能力也有明显增强。这可能归因于简单的浸渍无法使Ag(I)还原成Ag(0),而在光催化制氢过程中,Ag(0)才逐渐形成,与XRD表征结果一致;由于Ag/HNb3O8的表面等离激元效应,Ag/HNb3O8-DB和Ag/HNb3O8-PP-1在可见光区都产生了新的吸收峰,表明负载Ag对催化剂的能带结构产生了影响。
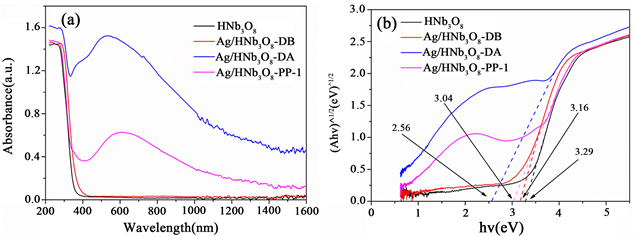
Figure 8. UV-vis-NIR spectra and Tauc plots of various samples
图8. 不同样品的紫外–可见近红外光谱和Tauc曲线图
合适的禁带宽度Ebg对催化剂的光催化活性有重要影响,它能决定入射光的截止波长。为了进一步探究催化剂禁带宽度对水裂解析氢的性能的影响,我们采用Tauc方法得到各催化剂的禁带宽度,如图8(b)所示。HNb3O8、Ag/HNb3O8-DB、Ag/HNb3O8-DA和Ag/HNb3O8-PP-1的禁带宽度分别为3.29、3.16、2.56和3.04 eV。水裂解反应需克服最小值为237 kJ∙mol−1的吉布斯自由能,等于1.23 eV势垒 [14] 。但是考虑到0.3~0.4 eV的内能损耗和0.4~0.6 eV的过电位要求,半导体的带隙必须大于1.8 eV [15] 。此外,吸收可见光能量的禁带宽度应小于3.2 eV。因此,用于水裂解的光催化剂的禁带宽度Ebg建议在1.8~3.2 eV之间。由各催化剂的禁带宽度可知,除纯HNb3O8纳米片外,其它催化剂的禁带宽度均在1.8~3.2 eV之间,因此负载Ag的HNb3O8纳米片对水裂解析氢反应表现出更好的光催化性能,并且Ag/HNb3O8-DA的禁带宽度最小,其光催化性能最好。
光催化剂的瞬态光电流响应与光生载流子的复合率密切相关。催化剂在一定波长的光的激发下,产生光生载流子,光生电子迁移过程中会产生光电流。因此,光电流越大,说明光生电子和空穴越难复合。图9为HNb3O8、Ag/HNb3O8-DA和Ag/HNb3O8-pp光催化剂在Xe灯照射下的瞬态光电流响应(PC)测试结果。当开灯或关灯时,所有样品都表现出良好的PC再现性。采用光沉积法负载Ag的光催化剂的光电流强度随着Ag负载量增多而增强,Ag/HNb3O8-pp-1材料表现出最高的PC强度;采用浸渍法负载Ag的催化剂Ag/HNb3O8-DA表现了更高的PC强度。这表明负载Ag(0)提高了光催化剂的电子–空穴对的分离能力 [16] [17] 。这与紫外–可见近红外光谱和光催化性能测试结果一致。
图9. 不同催化剂的瞬态光电流曲线图
光致发光光谱(PL)可以反映光催化剂中光生电子–空穴对的分离复合情况。因此,在激发波长为310 nm的条件下,对各催化剂进行了光致发光光谱(PL)测试。当催化剂受到一定波长的光激发后,基态电子吸收能量从低能级向高能级转移,并产生电子–空穴对。由于激发态电子不稳定,它们可以重新复合而发生荧光,这个过程会产生荧光发射峰。如图10所示,HNb3O8纳米片在波长为456 nm处产生最强的荧光发射峰,而其它负载Ag的催化剂也在此处产生最强的峰,但其荧光强度均有所下降,其中Ag/HNb3O8-DA催化剂的荧光发射峰最低。不同的荧光强度表示不同的电子–空穴复合程度。当荧光强度较高时,表明激发态电子经衰变回到基态的数量较大,表明电子复合速率较高。相反,随着荧光强度下降,表明催化剂的光生电子–空穴对的复合受到一定程度的抑制 [18] ,这个过程中,光生电子将移动到催化剂的表面。其形成原因可能是Ag的负载支持更多的活性位点,提高了催化剂的光生电子–空穴对的分离能力与电荷转移效率。这与光催化性能测试结果一致。
3.2. 催化剂的催化性能
为了探究各催化剂的催化析氢活性,我们测试了各催化剂在不同波长范围的光照射下的光催化析氢活性。图11(a)是各催化剂在紫外可见光(300 nm < λ < 780 nm)照射下的析氢量(μmol∙g−1)随时间(h)变化曲线。显然光沉积法制备的催化剂的光催化析氢活性随着Ag的增加而增强,当负载Ag的含量为1%时,催化剂活性最高。这可能是Ag(0)具有LSPR效应,较多的Ag(0)为光催化水裂解制氢反应提供了更多的反应活性位点,从而提高了光催化反应速率。用浸渍法制备的催化剂Ag/HNb3O8则表现出了更高的催化活性,由前面表征结果可知,Ag/HNb3O8具有合更强的LSPR效应,对可见–近红外光的吸收更强,因此表现出了更强的光催化活性。并且,析氢量随时间线性增加,这表明每小时氢气的生成都是均等的,且析氢速率相对稳定。
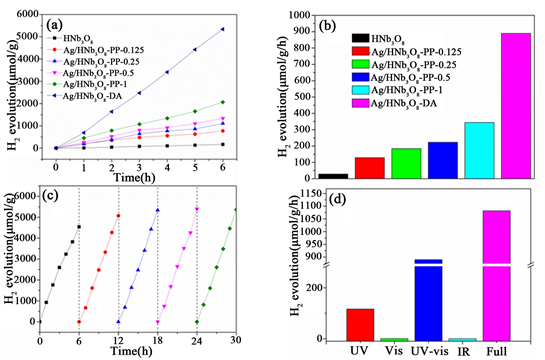
Figure 11. (a, b) Photocatalytic activity for hydrogen generation over various catalysts under UV-vis light; (c) Cycling measurement of Ag/HNb3O8-DAunder UV-vis light; (d) Photocatalytic activity for hydrogen generation over Ag/HNb3O8-DA under different light
图11. (a, b) 紫外可见光照射下,各催化剂的析氢活性;(c) 在紫外可见光照射下,Ag/HNb3O8
的光催化析氢的循环稳定性;(d) 不同光源照射下Ag/HNb3O8-DA的光催化析氢活性
为了探究各催化剂析氢速率的关系,我们测量了各催化剂每小时的析氢量,如图11(b)所示。HNb3O8的析氢量为28.4 μmol∙g−1h−1;Ag/HNb3O8-pp的析氢量随着Ag含量的增加而增大,其中Ag/HNb3O8-pp-1的析氢量最大,达到343.8 μmol∙g−1h−1,是HNb3O8的12.4倍;Ag/HNb3O8-DAAg/HNb3O8-DA的析氢量为889.7 μmolg−1h−1,是HNb3O8的31.3倍。这表明Ag具有较强的LSPR效应,该结果与催化剂的紫外可见近红外光谱表征结果一致。
图11(c)是在紫外可见光(300 nm < λ < 780 nm)照射下Ag/HNb3O8的循环稳定性测试结果。具体方法如下,反应时间6小时,每隔1小时测一次析氢量,反应后将催化剂用去离子水洗涤5次,收集样品,重复上述步骤5次。图可以看出,连续的5次析氢测试中,催化剂的析氢活性没有明显差异,说明制备的催化剂具较好的化学稳定性。在第1次析氢实验中,析氢量相对较低,第2~5次析氢实验中的析氢量略高于第1次析氢实验中的析氢量。这可能由于第一次光催化析氢测试时,Ag/HNb3O8-DB催化剂上吸附的大部分为Ag(I)而Ag(0)的含量很低,催化剂经过一次光催化反应后,Ag(0)的含量增加,使得Ag/HNb3O8催化剂在2~5次析氢反应中表现出更好的光催化活性,这与我们的XRD图谱和紫外–可见–近红外光谱的表征结果一致。
为了进一步探究Ag/HNb3O8-DA催化析氢活性,我们采用不同波长范围的光照射反应溶液进行了光催化析氢实验(图11(d))。仅在紫外光(300 nm < λ < 400 nm)照射下,HNb3O8-DA的析氢速率为116.9 μmol g−1h−1。在紫外–可见光(300 nm < λ < 780 nm)照射下,Ag/HNb3O8-DA的析氢速率为889.7 μmol∙g−1h−1,约为在紫外照射下HNb3O8-DA析氢速率的7.6倍。进一步向近红外光谱扩展,析氢速率增加了21.6%,达到1081.9 μmol∙g−1∙h−1。然而,Ag/HNb3O8-DA只在可见光(420 nm < λ < 780 nm)和近红外光(800 nm < λ < 2500 nm)照射下,几乎没有氢气生成(仅为0.8和0.1 μmol∙g−1∙h−1)。结果表明,虽然可见光和近红外光被Ag/HNb3O8-DA部分吸收,但所提供的能量不足产生光生电子–空穴对 [18] 。因此,在紫外–可见光和全光谱照射下,Ag/HNb3O8-DA的光催化析氢活性的增强不是产生了更多的光生电子–空穴对,而是催化剂对可见–近红外光的吸收提供了紫外光激发的光生电子–空穴对迁移所需要的额外动能,从而提高了Ag/HNb3O8-DA的光催化析氢活性。结合紫外–可见近红外结果,Ag/HNb3O8-DA纳米片表现出较强的LSPR效应。表面等离激元不仅可以产生高能热载流子,还可以提供热能来提高晶格温度,这都可以提高反应活性。
4. 结论
采用光沉积和浸渍的方法成功合成了负载Ag的HNb3O8纳米片Ag/HNb3O8-pp和Ag/HNb3O8光催化剂。研究了催化剂对水裂解析氢反应的光催化活性,在紫外可见光照射下,Ag/HNb3O8-DA表现出了最强的析氢活性。我们又研究了Ag/HNb3O8-DA在不同波长范围的光照射下的析氢活性,仅在紫外光照射下,Ag/HNb3O8-DA的析氢速率为116.9 μmol∙g−1∙h−1;在紫外可见光照射下,Ag/HNb3O8-DA的析氢速率为889.7 μmol∙g−1∙h−1,是紫外光照射下Ag/HNb3O8-DA析氢速率的7.6倍。进一步向近红外光谱扩展,析氢速率增加了21.6%,达到1081.9 μmol∙g−1∙h−1。PC和PL的表征结果进一步验证了Ag/HNb3O8-DA提高了光生电子–空穴对的分离能力,减少了电子–空穴对复合,从而提高了Ag/HNb3O8-DA光催化析氢活性。Ag/HNb3O8的LSPR可以有效地促进光诱导电子–空穴对(h+-e−)的分离和运输,并且提供了紫外光激发的光生电子–空穴对迁移所遇的额外动能。