1. 绪论
检测SF6分解组分的意义及原理
SF6因为其灭弧和绝缘性能相较于其他气体更加出色,所以被大量的运用在一些电气设备当中。气体绝缘组合电器(Gas Insulated Switchgear, GIS)就是将各种电气设备封装到一个带有接地装置的金属容器中,其外壳主要是由铝合金制造,用铝合金制造的外壳不仅可以增强其抗腐蚀的能力,而且可以降低电气设备本身的重量。GIS设备使用SF6作为其绝缘和灭弧介质,充入SF6的量约为0.4~0.6 MPa,GIS设备具有许多其他电气设备所没有的优点,如体积和重量都比较小,这使得其占地面积更小,GIS设备的占地面积仅仅达到户外变电站的30%,这可以非常有效的解决我国建筑用地紧张的问题。不仅如此,其安全可靠系数高。GIS设备密封性很强,绝缘性能几乎不随外界环境条件的变化而变化,以SF6作为绝缘介质其灭弧性能也很优异,当GIS设备内部发生开合闸时,电弧可以再非常短的时间内被熄灭,防止GIS设备内部的电弧重新燃烧,增强电力系统在输变电环节的安全可靠性 [1] 。
GIS设备具有稳定可靠、体积小、重量轻等优点,这也使得其日渐成为了电力系统输变电的重要组成成分,无论在国内还是国外都被广泛应用于电力传输,伴随着电力系统输变电环节技术的革新进步,投入运行的GIS设备的数量也是越来越多,GIS在电力传输环节的地位也是水涨船高。伴随着越来越多的GIS设备的使用,同时也凸显了以SF6为绝缘介质的GIS设备的绝缘缺陷,其在早期投入使用的过程中很难发现,但是GIS设备在长时间工作中也加快了其内部绝缘介质SF6老化的速度,伴随着使用时间的不断增加,并可能导致新的绝缘缺陷的产生,引起GIS设备内部发生局部放电(Partial Discharge, PD)或局部发热 [2] 。
如图1所示,GIS设备由于制造工艺的问题,在其内部难免出现一些金属凸起和毛刺等,这些由于现代制作工艺所带来的缺陷将会导致GIS设备随着使用时间的增加从而产生一些绝缘故障。这些缺陷导致了GIS设备内部的部分区域的局部电场强度变大进而引发局部放电,直接导致SF6受热分解产生性质不稳定的SF4等多种硫的低氟化物,等到温度降低时,以上分解产物发生可逆反应生成SF6气体 [3] 。但是实际工程所用的SF6气体并非是纯净的,通常含有极少量的氧气和水等副产物,其在高温环境中通常会分解生成性质非常不稳定的低氯化合物,并且低氯化合物在之后又会与SF6气体中存在的少量的氧气和水发生化学反应,从而生成化学性质更为稳定的CS2、SOF2、SO2F2等产物 [4] 。由于反应不可逆,所以这些产物无法还原生成SF6气体,从而导致GIS设备内部绝缘性能随着工作时间的增长变得越来越差,长时间过后必然会对设备的安全可靠稳定运行产生不好的影响。此外,由于产物多为含硫、含氟化合物,这些产物对GIS设备的金属材料和固体绝缘具有较强的腐蚀作用,不仅增加了设备的维护成本的负担,还给电力系统输送电环节的安全可靠带来了威胁 [5] 。
因此,利用Material Studios软件,探究SF6局部放电特征分解气体组分之一CS2分子,在N-TiO2上的吸附特性(吸附距离、电荷转移量、态密度、前沿分子轨道等),对GIS内部SF6分解气体组分在局部放电情况下进行测定,测出的分解组分可以用来作为气体绝缘开关设备内部存在缺陷的重要依据,对研制相应的智能电网SF6局部放电特征分解组分气敏传感器具有重要的工程和科学意义 [6] 。
2. 仿真实验原理及方法
密度泛函理论
密度泛函理论(Density functional theory, DFT)主要研究的是多个电子体系结构的一种量子力学计算方法。由于波函数的变量实在是太多,在实际的应用当中不方便处理,所以要用电子密度去取代波函数作为研究的基础变量,电子密度仅仅只是相当于三个变量的函数,其不管是在概念上还是实际应用中都要比波函数更加简单。
当Thomas-Fermi模型被建立的时候,密度泛函理论正式被世人所发现,但是那时的密度泛函理论在论据上还不够充分,所以在实际应用中也是颇受限制,后来由于Hohenberg-Kohn定理被提了出来,密度泛函理论才有了充分的理论依据作为支撑。密度泛函理论证明了当变量是基态密度的时候,便可以将整个体系的能量最小化,于是便得到了基态变量。密度泛函理论虽然有Hohenberg-Kohn定理作为其理论依据的支撑,但是其也只能计算体系的基态性能。
Hohenberg-Kohn定理:
(2.1)
(2.2)
Kohn-Sham方程:
(2.3)
基于DFT的自洽计算过程:
① 生成KS势:
(2.4)
② 求解KS方程:
(2.5)
③ 得到新的nout(r):
(2.6)
④ 与之前的nstart(r)比较收敛与否,最后再输出结果。
虽然固态物理学的计算方法有很多,但是密度泛函理论作为量子力学的一种计算方法,其在计算精度相较于其它方法也有其优势。由于固体物理学计算的精度要求非常高,而密度泛函理论的精度则是正好满足固体物理学的计算精度的要求。密度泛函理论主要是采用了与局域密度近似的计算方法,并且在固态计算中的花费要比实验更少,也为科研工作节省了不少的开支。虽然密度泛函理论的精度相比其它的物理学计算方法要好不少,但是随着时间的推移,人们对于物理学计算精度的要求也是越来越高,没有经过改进的密度泛函理论已经逐渐不能满足近乎苛刻的精度要求。
密度泛函理论子在物理学计算当中的应用越来越广泛,其也在不断的被科研工作者逐渐完善改进,但是它也有着一些局限性,如分子间相互作用和范德瓦尔斯力的计算与分析。
3. 仿真内容
3.1. 仿真计算方法
这一次的主要是在Materials Studio的Visualizer模块中分别建立TiO2和CS2分子的球棍模型(图2、图3),之后再用Dmol3模块对两者的模型进行结构优化,优化后的TiO2和CS2的球棍模型与实际的分子模型在结构误差上就可以忽略不计,并且可以让N-TiO2对CS2进行吸附实验得出数据与理论值的差值在实验所允许的误差以内。

Figure 2. Ball-and-stick model of the CS2 molecule
图2. CS2分子的球棍模型
 (a)
(a) 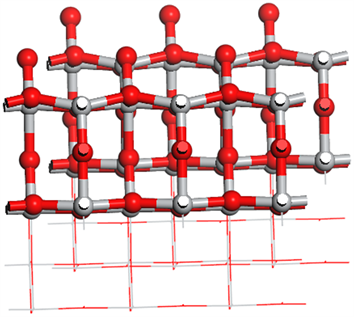 (b)
(b)
Figure 3. Different viewing angles of undoped TiO2 crystal planes
图3. 未掺杂的TiO2晶面的不同视角图
TiO2和CS2球棍模型建立及其吸附:
首先在Materials Studio软件中将TiO2和CS2的球棍模型建立起来,利用Dmol3模块对TiO2和CS2分子的球棍模型进行结构上的优化。再对TiO2进行氮原子掺杂,将TiO2微观模型中处于二配位的氧原子替换为氮原子,利用Materials Studio软件的Dmol3模块优化计算单个及两个CS2分子球棍模型靠近TiO2中的氮原子距离为2左右并且达到稳定后的结构。
3.2. 氮掺杂的TiO2模型构建
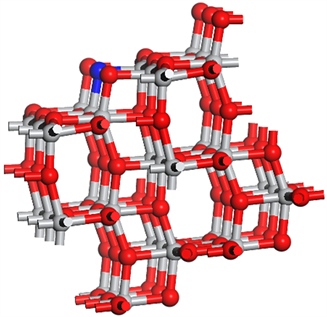
N-TiO2模型的构建主要是将通过MS软件的Dmol3模块结构优化好的TiO2上的氧原子由氮原子取代构成N-Ti键(图4)。
 (a)
(a)  (b)
(b)
Figure 4. Different viewing angles of N-doped TiO2 crystal planes
图4. N掺杂的TiO2晶面的不同视角图
在图5中对比了未掺杂氮原子的TiO2和N-TiO2的态密度,我们可以发现在两者的态密度对比图中N-TiO2的峰值要比TiO2的峰值高一些,在图6和图7能带结构图中可以得知未掺杂的TiO2的能隙为1.910 eV,而掺杂氮原子后的TiO2的能隙为1.906 eV,这表示TiO2在掺杂氮原子后的能隙宽度降低了,能隙宽度的降低也直接影响到了价带电子跃迁到导带的能力,其可以使得价带电子更容易跃迁到导带,并且之后吸附基底的导电性也会有所增强。

Figure 5. Comparison of the density of states of undoped TiO2 and N-TiO2
图5. 未掺杂的TiO2及N-TiO2的态密度对比图

Figure 6. The energy band structure of N-TiO2
图6. N-TiO2的能带结构

Figure 7. Band structure of undoped TiO2
图7. 未掺杂的TiO2的能带结构
3.3. Dmol3参数设置
Dmol3模块中的Setup选项卡中,task选择Geometry,Optimization,Energy (能量收敛精度)设置为1.0e−5 Ha,Max.force (能量梯度)设置为0.002 Ha/Å,Max.displacement (原子偏移)设置为0.005Å,Max.iterations数值为500,Max.step size数值为0.3Å,Quality选择Fine。选择泛函时选择GGA,主要是因为GGA(General Gradient Approximation,GGA)在广义密度上的精确度要比局域密度近似LDA (Local Density Approximation, LDA)高,之后再选择PBE泛函处理电子间的交换互联作用。之后再将Use勾选并选择Grimme,还需要勾选的有Spin unrestricted和Use formal spin as initial。
Dmol3模块中的Electronic选项卡中,Integration accuracy、k-point set选择Medium,SCF tolerance选择fine,core treatment选择All Electron,为了使得到的数据尽可能误差更小则选择Basis set时选择DNP,且在More选项中勾选Use DIIS和Use smearing;在K-points (布里渊k点网格)中选择Custom grid parameters后,Quality和Separation的数值将设为默认,Grid parameters设为1 × 1 × 1。
Dmol3模块中的Properties选项卡中,勾选Band Structure (能带结构)的同时设置Empty bands的数值为12,k-point set的精度设为Medium,Separation设为0.025;勾选Density of states (态密度)同时设置Empty bands的数值为12,Quality的精度设为Medium,并且勾选Calculate PDOS (局部态密度);勾选Electron density (电子密度)的同时将total density和Deformation density两个勾选;勾选Electrostatics (静电势)的同时对Electrostatic potential勾选;勾选opulation analysis (布居分析)的同时将Mulliken analysis、Hirshfeld analysis分别设置为Atomic Change和Charge。
3.4. 单个及两个CS2分子在N-TiO2表面的吸附
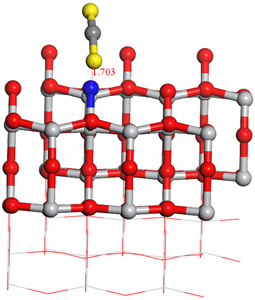
为了分析不同个数的CS2分子在氮掺杂的TiO2晶体表面进行吸附时对其的气敏特性的影响,所以必须做两次吸附实验,即分别进行单个CS2分子和两个CS2分子吸附氮掺杂的TiO2的仿真实验(图8),单个CS2和两个CS2分子吸附N-TiO2的吸附距离在2和3之间即可,并且在Dmol3模块中将需要的参数全部设置好即可开始吸附仿真实验。当吸附仿真实验成功时,就可以进行数据的分析和相应的图表的绘制,其中主要的数据有吸附距离、吸附能、电荷转移量、能带结构、态密度(图9)。
 (a)
(a) 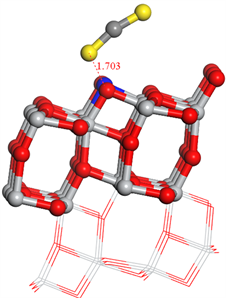 (b)
(b) 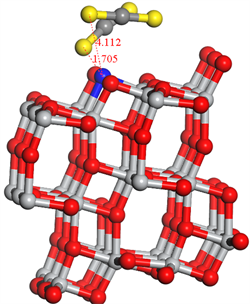 (c)
(c)  (d)
(d)
Figure 8. Different viewing angles of single and double CS2 adsorbed N-TiO2 crystal faces stabilized
图8. 单个及两个CS2吸附N-TiO2晶面稳定后的不同视角图
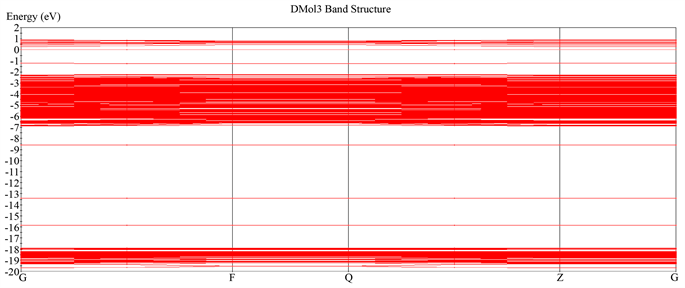
Figure 9. The energy band structure of single CS2 adsorbed N-TiO2
图9. 单个CS2吸附N-TiO2的能带结构
吸附能Ea表示的是CS2分子在氮掺杂的TiO2表面进行吸附前后其总能量的改变量,因此我们可以根据Ea绝对值的大小来判断CS2分子在氮掺杂的TiO2晶体表面进行吸附的能力强弱,其公式:
(3.1)
吸附能Egas表示的是一个CS2分子的能量值,Esur表示的是氮掺杂的TiO2的能量值,Esur+gas表示的就是硫化物分子在氮掺杂的TiO2表面进行吸附仿真实验结构稳定后的能量值。Ea < 0即表示CS2在吸附氮掺杂的TiO2并且结构达到稳定后整个过程中是在释放能量,反之Ea > 0即表示CS2在吸附氮掺杂的TiO2并且结构达到稳定后整个过程中是在吸收能量的,Ea的绝对值越大就表示CS2在吸附氮掺杂的TiO2的过程中释放或者吸收的能量就越多,那么CS2与氮掺杂的TiO2的吸附作用反应就越强烈,最后形成的结构就越稳定。
表1中主要包含了吸附能、电荷转移量和稳定距离这三个关键变量,为了弄清楚CS2在吸附氮掺杂的TiO2晶体表面结构达到稳定的过程中的电荷转移量,因此在整个吸附仿真的实验过程中通过计算得到了电荷转移量。计算电荷转移量的主要目的是为了了解在整个仿真吸附实验的过程中CS2分子的电子的走向,如果电荷转移量Q < 0,那么便表示CS2在吸附N-TiO2晶体表面且结构达到稳定过程中的电荷转移是从N-TiO2到CS2,反之电荷转移量Q > 0则表示电荷转移是从CS2到N-TiO2。电荷转移量Q的含义便是CS2分子在N-TiO2晶体表面吸附结构达到稳定后的与CS2分子进行吸附仿真实验之前的电荷分布的变化值,表1中单个CS2分子吸附N-TiO2晶体且结构稳定后的电荷转移量Q为−0.001e,即表示在整个吸附过程中电荷转移是从N-TiO2到CS2分子,N-TiO2失去电子;两个CS2分子吸附N-TiO2晶体且结构稳定后的电荷转移量Q为0.005e,即表示在整个吸附过程中的电荷转移是从CS2分子到N-TiO2,CS2分子失去电子。

Table 1. The data variables after the crystal structure of N-TiO2 is stabilized by adsorption of single and two CS2 molecules
表1. 单个及两CS2分子吸附N-TiO2晶体结构稳定后的数据变量
单个CS2分子和两个CS2分子在N-TiO2晶体表面进行吸附仿真实验过程中的吸附能分别为20.355 Ha和20.333 Ha,两者在吸附N-TiO2的整个过程中都是吸收能量,并且数值上差别可以忽略不计。电荷转移量分别为−0.001e和0.005e,两者在进行吸附仿真实验最后结构达到稳定后其S原子与N-TiO2晶体中的氮原子的距离分别为1.703Å和1.705Å、4.112Å。由上述数据可以得知无论是一个还是两个CS2分子进行吸附仿真实验过程中都是吸收能量。
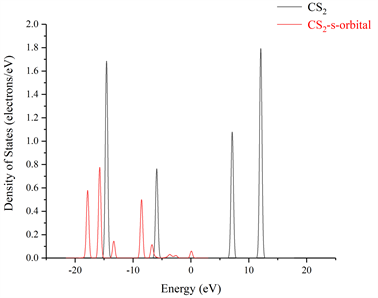
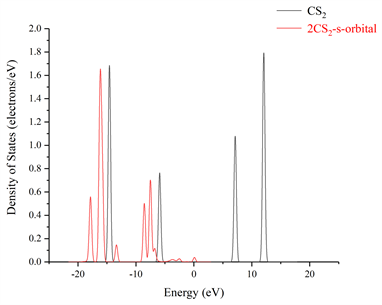
图10所示单个CS2分子在吸附氮掺杂的TiO2且结构稳定后的吸附体系的总态密度(Total Density of State,PDOS),其中CS2-S-TiO2表示的是用一个CS2分子中的硫原子靠近氮掺杂的TiO2晶面中的氮原子;图11所示两个CS2分子吸附氮掺杂的TiO2且结构稳定后的吸附体系的总态密度,2CS2-S-TiO2表示的是用两个CS2分子中的S原子靠近氮掺杂的TiO2晶面中的氮原子。图10和图11可以看出不论是单个还是两个CS2分子在吸附N-TiO2晶体且结构稳定的过程中,CS2分子的态密度在整个吸附体系的费米能级附近均有分布,并且相较于单个CS2分子吸附N-TiO2晶体结构稳定后的s或p轨道的导带态密度,两个CS2分子吸附N-TiO2晶体结构稳定后的导带态密度明显要比前者多,这说明两个CS2在吸附N-TiO2晶体且结构达到稳定后对整个吸附体系的导带态密度贡献更大;而且我们从图12和图13也可以发现不论是单个或两个CS2分子,两者在吸附N-TiO2且结构稳定后的s和p轨道的态密度的峰值都要比吸附前的CS2分子的s和p轨道的态密度峰值小,不仅如此,单个和两个CS2分子在吸附N-TiO2晶体且结构达到稳定后的X轴正半轴s和p轨道的态密度已经没有了波形。
图14可以发现两个CS2分子吸附N-TiO2晶体且结构达到稳定后的态密度峰值相较于单个CS2分子吸附N-TiO2晶体结构稳定后的态密度要大,两个CS2分子结构稳定后整个吸附体系在X轴的−16、−14、−8、−7.5 eV等坐标位置的态密度峰值都要比单个CS2分子结构稳定后的态密度峰值大,并且单个CS2分子吸附结构稳定后的吸附体系的态密度波形在X轴正半轴就已经没有了。
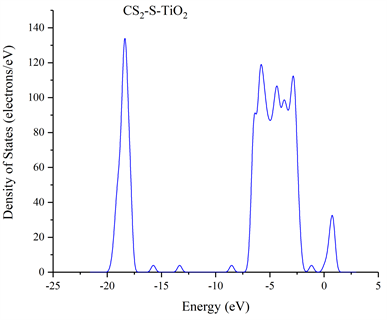
Figure 10. The total density of states of the adsorption system after the adsorption of a single CS2 molecule to stabilize the structure of N-TiO2
图10. 单个CS2分子吸附N-TiO2结构稳定后吸附体系总态密度
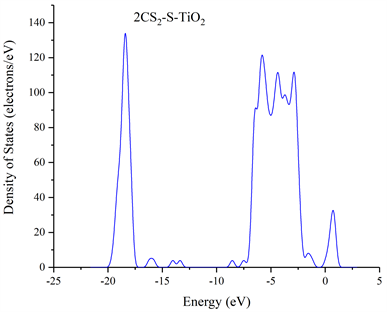
Figure 11. The total density of states of the adsorption system after the adsorption of two CS2 molecules to stabilize the structure of N-TiO2
图11. 两个CS2分子吸附N-TiO2结构稳定后吸附体系总态密度
 (a)
(a)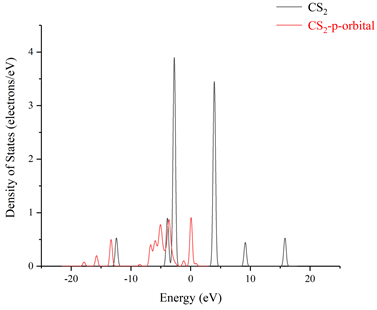 (b)
(b)
Figure 12. The comparison of the density of states of the s and p orbitals of CS2 after the structure of N-TiO2 is stabilized by the adsorption of single CS2 and before the adsorption
图12. 单个CS2吸附N-TiO2结构稳定后与未吸附前CS2的s和p轨道的态密度对比图
 (a)
(a)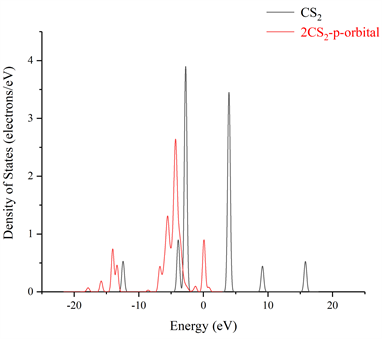 (b)
(b)
Figure 13. The comparison of the density of states of the s and p orbitals of CS2 after two CS2 adsorbed N-TiO2 structures stabilized and before the adsorption
图13. 两个CS2吸附N-TiO2结构稳定后与未吸附前CS2的s和p轨道的态密度对比图

Figure 14. The total density of states of the adsorption system after the adsorption of single and two CS2 molecules to stabilize the structure of N-TiO2
图14. 单个及两个CS2分子吸附N-TiO2结构稳定后吸附体系总态密度
4. 结论与展望
本文基于密度泛函理论,对SF6分解气体在N掺杂TiO2上的吸附进行了理论计算。为了全面了解这些吸附过程,考虑了Mulliken布居分析、DOS和前沿分子轨道理论。具体结论如下:
1) 单个CS2分子吸附N-TiO2晶体且结构稳定后的电荷转移量Q为−0.001e,N-TiO2晶面的导电性有所降低;两个CS2分子吸附N-TiO2晶体且结构稳定后的电荷转移量Q为0.005e,N-TiO2晶面的导电性有所上升。不论是单个CS2分子还是两个CS2分子在吸附N-TiO2晶体且结构稳定后的电荷转移量都可以忽略不计。
2) 单个CS2分子和两个CS2分子在N-TiO2晶体表面进行吸附仿真实验过程中的吸附能分别为20.355 Ha和20.333 Ha,这说明两者在吸附N-TiO2晶体的过程中都是吸收能量;并且两者数值差别几乎可以忽略,这说明N-TiO2晶体吸附CS2分子能力的强弱与被吸附的分子数量关系不大。
3) 两个CS2分子吸附在N-TiO2晶体上的能隙相较于单个CS2分子吸附后稳定的体系的能隙要小,这表明两个CS2分子的吸附体系比单个分子的吸附体系的价带电子更容易跃迁到导带,并且之后吸附基底的导电性能也有所增强。