摘要:
目的:探讨儿童吉兰–巴雷综合征变异型的临床特点、诊断和治疗。方法:分析青岛大学附属医院儿科2例吉兰–巴雷综合征变异型患儿的临床表现、相关辅助检查结果以及临床治疗效果。结果:临床表现为言语含糊、饮水呛咳、吞咽困难,外周神经病神经节苷脂抗体分别为抗GM1_IgM抗体阳性及抗GM4 IgG抗体阳性,脑脊液检查未见明显蛋白–细胞分离现象。肌电图示神经源性损害。2例均给予静滴免疫球蛋白治疗后症状明显好转。结论:急性延髓麻痹是吉兰–巴雷综合征的少见变异型,极易出现漏诊和误诊,须结合临床表现、脑脊液、电生理和影像学检查及早做出诊断和治疗。
Abstract:
Objective: To explore the clinical feature, diagnosis and treatment of the variants of Guillain-Barre syndrome (GBS) in Children. Methods: The clinical manifestations, relevant auxiliary examination results and clinical therapeutic effects of two children with GBS variants from Department of Pediatrics, the Affiliated Hospital of Qingdao University were analyzed. Results: The clinical manifestations included speech disorder, bucking because of drinking water, and difficulty in swallowing. The ganglioside antibodies of peripheral neuropathy were anti-GM1 IgM antibody positive and anti-GM4 IgG antibody positive. Cerebrospinal fluid examination showed that there was no obvious protein-cell isolation. Electromyography indicated that there were neurogenic damages. The symptoms of the two children improved significantly after they were given intravenous drip of immune globulin. Conclusion: Acute bulbar paralysis is a rare variant of GBS, so misdiagnosis and missed diagnosis are very common. Early diagnosis and treatment of acute bulbar paralysis should be done by combining clinical manifestations, cerebrospinal fluid, electrophysiology and imaging examination.
1. 序言
吉兰–巴雷综合征(Guillain-Barre Syndrome, GBS)是一种免疫介导的急性单相多神经根神经病,主要损害脊神经根和周围神经,也可累及神经系统其它部位 [1]。随着对疾病的深入认识和临床实践,越来越多的不典型或变异型GBS被广泛报道,不断打破了以往的诊断标准。GBS临床表现的多样化与不同神经根、周围神经及脑干的选择性受累有关 [2]。吉兰–巴雷综合征的变异型较少见,症状、体征表现多样,诊断困难,须与重症肌无力、脊髓灰质炎、急性播散性脑脊髓炎、多发性周围神经病、中毒性神经病变、周期性麻痹、血卟啉症和代谢紊乱等相鉴别 [3] [4]。本文介绍2例儿童变异型GBS,以提高临床对该病的认识。
2. 病例资料
病例1:患儿,男,8岁,因言语含糊伴饮水呛咳3天入院。发病前有上呼吸道感染史。患儿3天前出现言语含糊,说话缓慢、费力,伴饮水呛咳,进食固体食物时无呛咳,无晨轻暮重,无肢体无力,无行走不稳,无麻木,无感觉障碍,无发热,无咳喘,无呼吸困难,无吐泻,无抽搐,无尿便障碍。当地医院考虑诊断“咽炎”,抗感染治疗症状无缓解,无进行性加重。体格检查:体温36.1℃,脉搏80次/min,呼吸20次/min,血压116/71 mmHg;心肺腹检查未见明显异常。神经系统检查:神志清,高级智能活动正常。双眼球各方向运动自如,双侧瞳孔直接及间接对光反射存在,鼓腮无漏气,示齿右侧鼻唇沟变浅,伸舌偏右,咽反射消失。肌力肌张力正常。腱反射正常。入院前1天当地行颅脑CT检查无异常。入院第2天新斯的明试验阴性,血常规、生化、抗核抗体、降钙素原、血沉、大小便常规均无异常,脑脊液常规、生化、免疫球蛋白、细菌涂片、细菌培养均无异常。自身免疫性周围神经病:抗GM1IgM抗体阳性。颅脑MR平扫成像 + DWI成像、脑动脉MR血管成像(MRA)均无异常。脑静脉MR血管成像(MRV):右侧横窦、乙状窦管腔纤细,考虑发育性可能性大(见图1)。入院第3天电子纤维喉镜检查:双侧披裂略红肿,余无异常(见图2)。入院第4天颅脑(颅神经) MR平扫 + 单脏器薄层扫描:未见明显异常。入院第10天行肌电图检查:左侧上下肢周围神经、瞬目反射未见明显异常。诊治经过:综合上述检查,诊断吉兰–巴雷综合征急性延髓麻痹变异型。给予丙种球蛋白免疫封闭治疗,布地奈德混悬液雾化吸入治疗、甲钴胺营养神经治疗。入院第13天患儿言语渐恢复正常水平,无饮水呛咳,鼻唇沟对称,伸舌居中,咽反射恢复正常。

Figure 1. There is transverse sinus at the right side and the lumen of sigmoid sinus is thin, so high possibility of growth shall be considered
图1. 右侧横窦、乙状窦管腔纤细,考虑发育性可能性大

Figure 2. Arytenoid at both sides is a little red and swollen, and others are normal
图2. 双侧披裂略红肿,余无异常
病例2:患儿,男,7岁,因吞咽无力、咳嗽1周,声音改变6天入院。患儿1周前患上呼吸道感染后出现吞咽无力,进食固体食物梗阻感,须反复饮水方可缓解,喝水易从鼻腔流出,咽部不适,无饮水呛咳,偶咳嗽,6天前声音改变,鼻音明显。无晨轻暮重,无肢体无力、走路不稳及肢体麻木,无发热及呼吸困难,无吐泻、抽搐,无尿便障碍。曾就诊于当地医院行颅脑CT未见异常,考虑“软腭瘫痪”,给予口服“醋酸泼尼松5 mg qd,甲钴胺0.5 g qd”治疗5天,进食梗阻感稍有减轻,鼻音无缓解。随后就诊于我院,行颅脑MRI增强检查未见明显异常。体格检查:体温36.8℃,脉搏88次/min,呼吸22次/min,血压105/62 mmHg;心肺腹检查未见明显异常。神经系统检查:神志清,高级智能活动正常。双眼球各方向运动自如,双侧瞳孔直接及间接对光反射存在,鼓腮无漏气,伸舌居中,咽反射减弱。肌力肌张力正常。腱反射正常。入院第2天查血常规、生化全套、尿便常规均无异常。电子纤维喉镜示:鼻咽部淋巴组织增生,阻塞后鼻孔处1/2,会厌谷、梨状窝光滑,未见占位病变。双披裂运动好,双声带前中1/3交界处伪膜,声带运动对称、闭合可。入院第6天行上消化道X线造影示未见明显器质病变。新斯的明试验阴性。入院第8天行颅脑(颅神经) MR平扫 + 单脏器薄层扫描示:右侧舌咽神经、迷走神经及副神经根部局限性增粗;右侧小脑下前动脉返折处与右侧面听神经分界不清(见图3)。入院第11天脑脊液常规、生化、细菌涂片、免疫球蛋白均无异常;外送自身免疫性周围神经病示抗GM4IgG抗体阳性。入院第13天行肌电图检查:神经源性损害(见图4)。诊治经过:综合上述检查,诊断:吉兰巴雷综合征急性延髓麻痹变异型。给予丙种球蛋白免疫封闭治疗。入院第18天患儿吞咽无力及进食梗阻感明显减轻,鼻音消失,无声音改变,无咳嗽,咽反射恢复正常。
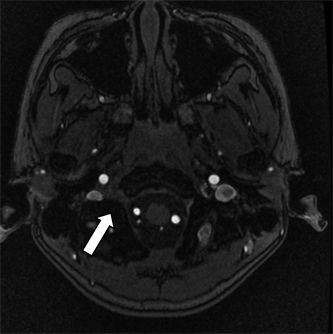
Figure 3. The root of glossopharyngeal nerve, vagus nerve and accessory nerve at the right side is locally thickening. (suggesting that there is inflammatory edema in the cranial nerves IX, X and XI sent by medulla oblongata)
图3. 右侧舌咽神经、迷走神经及副神经根部局限性增粗。(提示由延髓发出的第IX、X、XI对颅神经的炎性水肿)
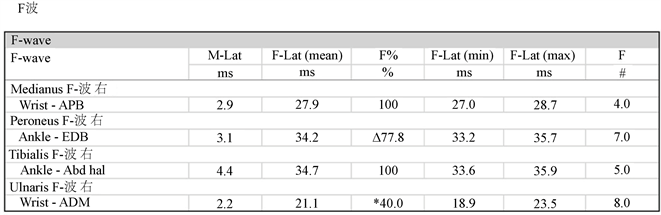
Figure 4. The MCV and SCV of ulnar nerve at the right side are both normal, F wave latency is normal, but its occurrence rate is decreased; The MCV of common peroneal nerve at the right side is normal, F wave latency is normal, but its occurrence rate is decreased
图4. 右侧尺神经MCV,SCV均正常,F波潜伏期正常,出现率降低;右侧腓总神经MCV正常,F波潜伏期正常,出现率降低
3. 文献复习
急性延髓麻痹(Acute Bulbar Palsy, ABP)可出现在各种神经系统疾病中,包括血管炎、重症肌无力、肉毒中毒和多发性硬化症等 [5]。2016年韩国一项多中心研究 [6] 提出将无肢体无力的急性延髓麻痹(ABP)作为GBS的一个变异型,其突出的临床特点为急性延髓麻痹而不伴有肢体无力。该研究指出,ABP依据临床表现可分为两类:急性延髓麻痹孤立型(ABPi)和急性延髓麻痹叠加型(ABPp)。ABPi仅表现为急性延髓麻痹;ABPp除了急性延髓麻痹外还可能伴发以下GBS的相关特征 [7]:其他颅神经受累(眼肌麻痹多见),感觉性共济失调,腱反射消失,脑脊液蛋白–细胞分离,血清抗GM抗体阳性,轻微的神经传导异常,其中眼肌麻痹和感觉性共济失调是最常见的伴随症状。无肢体无力的ABPp可以表现为MFS或PCB变异型中的各种临床表现的组合,但它不同于MFS和PCB的是,其延髓麻痹是整个疾病过程中最突出的症状,甚至在某些情况下也是最初的症状。我们的2例患儿临床上仅有延髓麻痹症状,有肌电图或是颅神经MRI的支持证据,随访再没有出现别的症状,排除其他病因,符合GBS的ABP变异型诊断。且2例患儿经IVIG治疗后,症状明显改善。故临床遇到急性延髓麻痹的病人,要想到可能为吉兰-巴雷综合征变异型,以期早期诊断及治疗。
以往研究表明,GBS的ABP变异型患者血清抗GT1a-IgG抗体阳性率最高,其次为抗GQ1b-IgG抗体。GT1a神经节苷脂存在于人类神经组织中,包括大脑、延髓、舌下神经核、下橄榄核和周围神经。GQ1b多分布于动眼神经、滑车神经、展神经的郎飞结和神经肌肉接头、四肢肌梭。抗GT1a抗体多与延髓麻痹相关,当与GQ1b抗体共存时则与眼肌麻痹高度相关 [8]。有趣的是,我们的患儿例1抗GM1IgM抗体阳性,例2抗GM4 IgG抗体阳性,这与以往报道不同。GM1在神经系统中含量丰富,是唯一可以透过血脑屏障的神经节苷脂 [9]。GM1抗体结合于前根运动神经轴突膜及郎飞氏结上的GM1,直接影响Na+通道的功能,导致动作电位传导受限,产生乏力症状。抗GM1抗体是GBS中最常见的抗神经节苷脂抗体,主要分布于外周运动神经轴突膜上,尤其是郎飞氏结。而抗GM4抗体与GBS相关性目前国内外研究甚少,具体发病机制不明。该2种抗体与ABP的相关性,还需进一步积攒病例,后续研究。
4. 讨论
综上所述,GBS的临床表现复杂且多样,可由感染性、炎性或全身性疾病引发。需要通过综合分析做出诊断。若符合脊神经和颅神经支配肌肉的对称性无力、单相病程(发病4周内达高峰期),即可考虑 GBS的诊断。腱反射减弱和/或消失、脑脊液蛋白–细胞分离现象已不再作为诊断的必备条件。神经传导检查可为GBS的诊断提供有力的支持证据,尤其是对非典型GBS的诊断。远端潜伏期延迟、神经传导速度减慢、波形时间弥散、传导阻滞、F波延长或缺失、H反射延长或缺失均是支持GBS周围神经脱髓鞘的表现 [10]。本文2例报道提醒我们临床上遇到ABP患者时,在排除其他疾病后,要想到GBS的ABP变异型,在临床工作中需要密切监测,因为他们有发展为急性呼吸衰竭的风险 [11]。我们要通过详细问诊、查体以及借助相关的实验室手段,做到早期诊断、早期治疗,绝大多数患者预后良好。
NOTES
*第一作者。
#通讯作者。