1. 引言
氧离子导体在氧泵、氧传感器和固体氧化物燃料电池领域都有着广阔的应用前景,因此是目前重要的研究材料之一。目前已经广泛地认识到钙钛矿型氧化物具有高的氧离子扩散率。近年来Sr、Mg掺杂的LaGaO3材料由于比传统的氧离子导体氧化钇稳定氧化锆(YSZ)具有更高的氧离子电导和宽的氧分压范围内的稳定性引起了人们的广泛注意 [1] - [6],有望成为SOFC固体电解质的理想材料之一。
作为SOFC的电解质材料,要求其氧离子电导尽可能高而电子电导尽可能低甚至可以忽略。氧离子电导主要由氧空位形成能和迁移能决定,与晶体结构有着密切的关系。然而,目前文献报道La0.9Sr0.1Ga0.8Mg0.2O3−δ在室温下有立方相 [1] [2]、正交相 [3] [4] 和单斜相 [5] [6] 三种晶体结构。Feng [1] 和Trofimenko [2] 证实LSGM在室温下为立方结构,空间群
(No. 221);Lerch et al. [4] 通过粉末中子衍射分析得出LSGM在室温下为正交结构(空间群Imma,No. 74),而在1073K为立方结构(空间群
,No. 221)的结论。Slater et al. [5] 则发现了更加复杂的结构变化,他们认为La0.9Sr0.1Ga0.8Mg0.2O3−δ在室温下具有单斜结构(空间群I2/a,No.15),在723 and 1273 K温度范围发生两相转变:单斜结构(假正交)-单斜结构(假斜方六面体)-斜方六面体结构(R3c)。因此,有关La0.9Sr0.1Ga0.8Mg0.2O3−δ晶体结构的问题目前仍未解决,没有形成一致性的结论。这种晶体结构的不确定性使得有关该材料氧离子电导和晶体结构内在关系的研究也变得困难重重。
基于密度泛函理论(DFT)的第一性原理计算是从细节上理解电子和氧空位输运机制的重要手段 [7] - [14]。人们已经针对LaGaO3基氧离子导体进行了许多模拟计算来研究其缺陷形成能 [7] 和氧空位迁移能 [8],这些前期的工作成功地再现了实验观察到的趋势。本文通过密度泛函理论计算系统地研究了立方、斜方六面体、正交和单斜结构LSGM的氧空位形成能和迁移能,目的是为了阐明不同晶体结构LSGM氧离子电导的内在机制。
2. 计算方法
我们采用了基于密度泛函理论(DFT)的DMol3计算程序包。在Dmol3软件包中所提供的LDA泛函有VWN和PWC两种,所提供的GGA泛函有PW91、BP、BOPBLYP、VWN-BP、PBE、RBPE和HCTC等几种。根据其他类似钙钛矿材料的文献报道 [7] [9],我们选用广义梯度近似(GGA)中的Perdew-Wang (PW91)泛函来描述电子交换相关能。DFT半核赝势(DSPP)是全电子计算中的局域赝势,其核内电子的效应由包含一定程度相对论效应的简单势来描述。对于Sr、Mg在LaGaO3中少量掺杂我们采用虚晶近似(VCA)的方法来建模,虚晶近似的方法至少在原理上能够用于研究和预言掺杂作用,并且已经证明对于钙钛矿固溶体是此种方法的研究是准确的。VCA计算是根据占有率的大小来确定赝势的贡献比例的。不同晶体结构的分数原子坐标和原子间距来源于文献报道 [4] [5],在本章的研究中共涉及立方(C-LSGM)、斜方六面体(R-LSGM)、正交(O-LSGM)和单斜(M-LSGM)四种晶体结构。为了达到绝对能量收敛(≤ 0.5 meVatom−1),基于Monkhorst-Pack scheme,四种晶体结构原胞中布里渊区k点的选取分别为C-LSGM:6 × 6 × 6 (10个不可约点),R-LSGM:4 × 4 × 4 (32 个不可约点),O-LSGM:4 × 5 × 4 (40个不可约点)和M-LSGM:4 × 4 × 4 (32个不可约点)。每种晶体结构原胞的晶格参数和内部坐标都按照残余力residual force ≤ 0.1 eVÅ-1的收敛标准进行充分优化。
含有一个氧空位的超胞用充分优化的四种原胞模型C-LSGM、R-LSGM、O-LSGM 和M-LSGM构建,超胞大小分别为C-LSGM:2 × 2 × 4,R-LSGM、O-LSGM和M-LSGM:2 × 2 × 2,每个含有氧空位的超胞都包含79个原子。超胞能量收敛标准与原胞相同(≤ 0.5 meVatom−1),k点的选取分别为C-LSGM:3 × 3 × 1,O-LSGM:2 × 3 × 2,R-LSGM和M-LSGM:2 × 2 × 2,超胞的结构弛豫限制在一个以缺陷位置为中心半径为3.2 Å的球形区域。对于气态氧分子总能量的计算,O2分子被放置在一个与O-LSGM超胞尺寸相同的真空盒子中,k点设置为2 × 2 × 2 (4个不可约点),同时需要考虑自旋极化。我们采用DMOL3程序包中的Complete LST/QST (Linear Synchronous Transit/Quadratic Synchronous Transit)方法来搜索过渡态,此方法能够以一种简单的方式显示过渡态并能够直接得到氧空位氧空位迁移势垒(Em)。
3. 计算模型及结构参数
图1(a)~(d)分别显示了立方(
)、斜方六面体(R3c)、正交(Imma)和单斜(I2/a)结构LSGM原胞的球棍模型图。
是一个完美的立方钙钛矿结构,R3c、Imma和I2/a则是由共顶角的GaO6八面体组成的,属于旋转扭曲的钙钛矿家族。

Figure 1. Ball-and-stick primitive cell models of LSGMs with different crystal symmetries (the grey, white and black spheres represent La/Sr, Ga/Mg and O ions, respectively): (a) C-LSGM (
); (b) R-LSGM (R
3 c
); (c) O-LSGM (Imma); (d) M-LSGM (I2/a)
图1. 不同晶体对称性LSGM原胞的球棍模型(灰、白和黑色球分别代表La/Sr, Ga/Mg和O离子):(a) C-LSGM (
);(b) R-LSGM (R3c);(c) O-LSGM (Imma);(d) M-LSGM (I2/a)
在计算氧空位之前,不同晶体对称性的LSGM原胞都通过能量最小化方法进行优化模拟,优化后的晶格参数列在表1中,并且和对应的实验值进行了对比,计算晶格参数与实验值非常接近,说明了后面的缺陷模拟是可靠的和有效的。

Table 1. Calculated and experimental structural parameters of cubic, rhombohedral, orthorhombic and monoclinic LSGM
表1. 立方、斜方六面体、正交和单斜四种结构LSGM计算和实验的结构参数
4. 结果及讨论
4.1. 氧空位的形成能
在Sr、Mg掺杂的LaGaO3 (LSGM)系统中主要的输运机制是氧空位的迁移,氧空位的形成主要通过如下缺陷反应产生:
其中
代表氧空位,
代表晶格氧。电导活化能总体上包括氧空位的形成能和迁移能。
氧空位的形成能通过如下方程计算得到:
其中
表示每个包含一个氧空位的超胞总能量计算值,
表示氧气分子中一个氧原子能量计算值,
则对应于相同超胞的完美晶体总能量计算值。
图2显示了计算得到立方、斜方六面体、正交和单斜四种结构LSGM中一个氧空位的形成能,在立方和斜方六面体结构LSGM中氧空位的位置仅有一种情况,因为所有的氧原子位置是等价的,而在正交和单斜结构LSGM中由于存在两种不等价的氧原子位置(O1和O2位),因此对应有两个氧空位形成能,其中O2位的形成能高于O1位,但是差别很小,仅有0.06~0.07 eV。图2显示立方结构LSGM的氧空位形成能为3.96 eV,远远小于斜方六面体、正交和单斜结构,后三种结构LSGM的氧空位形成能大约为5.50 eV,这意味着立方结构LSGM中的氧离子或许更容易被激发解离出来参与输运过程。
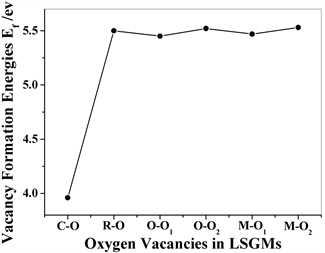
Figure 2. Calculated vacancy formation energies (Ef) of LSGMs with respect to the calculated energy (1/2)EO2 for an oxygen atom in the molecule. C-O and R-O corresponds to oxygen vacancy in cubic and rhombohedral LSGM, respectively. O-O1, O-O2, M-O1 and M-O2 correspond to oxygen vacancy at O1 or O2 site in orthorhombic and monoclinic LSGM, respectively
图2. 计算不同晶体结构LSGM的氧空位形成能(Ef),其中氧原子能量(1/2)EO2从氧气分子计算得到。C-O和R-O对应于立方和斜方六面体结构中氧空位,O-O1, O-O2, M-O1和M-O2则对应于正交和单斜结构中O1或O2位的氧空位
4.2. 氧空位的迁移能
对于立方和斜方六面体结构LSGM来讲,仅需要考虑一种空位迁移路径,而对于正交和单斜结构则需要考虑O1-O2和O2-O2两种空位迁移路径。在正交和单斜结构中由于两个最近邻的O1位距离太远(超过3.9 Å),因此两个O1位之间的迁移是不可能出现的 [10],这里勿需考虑。
我们使用DMOL3程序包中的complete LST/QST方法来搜索过渡态(TS),首先使用同步线性转变最大化[linear synchronous transit (LST)], 再沿着能量反应路径共轭的方向使用能量最小化的方法,然后对近似的过渡态再使用二次同步转变[quadratic synchronous transit (QST)]最大化。在此基础上另外一种方法——共轭梯度最小化被采用,此循环周而复始直到找到静态点,就这样搜索到过渡态并直接得到氧空位迁移的能量势垒。

Figure 3. Calculated vacancy migration energies (Em) of LSGMs along different migration pathways, C-O-O and R-O-O corresponds to the migration pathways of oxygen vacancy between two neighboring oxygen sites in cubic and rhombohedral LSGM, respectively. O-O1-O2, O-O2-O2, M-O1-O2 and M-O2-O2 correspond to the migration pathways of oxygen vacancy between neighboring O1 and O2 sites or two O2 sites in orthorhombic and monoclinic LSGM, respectively
图3. 四种结构中沿着不同迁移路径的氧空位迁移能(Em),C-O-O和R-O-O分别对应于立方和斜方六面体结构LSGM中两个近邻氧位之间的空位迁移能,O-O1-O2、O-O2-O2、M-O1-O2和M-O2-O2分别对应于正交和单斜结构LSGM中O1和O2位之间及两个近邻O2位之间的空位迁移能
对于立方和斜方六面体结构LSGM来说,氧离子在两个等价的氧空位之间发生迁移,对于正交和单斜结构LSGM来说,氧离子可以在两个近邻的O2位或者O1和O2位之间发生迁移。图3显示了计算得到四种结构中沿着每个迁移路径氧空位的迁移能。从图中可以清楚地看出立方结构LSGM的氧空位迁移能要远低于其他三种结构,迁移能按如下顺序增加
(
和
为沿着O1-O2和O2-O2两种迁移路径的平均值)。不同结构的空位迁移能出现一个明显的趋势:晶格越扭曲(对称性越低),迁移能就越大,这与文献报道其他钙钛矿材料的结论一致 [8] [11]。同时在正交和单斜结构LSGM中两个近邻O2位之间的空位迁移能要明显低于O1和O2位之间的迁移能,这表明在低温区氧空位的迁移在两个近邻O2位之间要比O1和O2位之间更容易发生,也有可能仅有前一种迁移路径发生迁移。
根据以上计算结果,立方结构LSGM中空位形成能和迁移能都远远小于其他三种结构,因此可以推测立方结构LSGM在四种结构中具有最高的氧离子电导。氧离子电导按如下顺序增加
,也就是说出现一个明显的趋势:晶格越接近完美立方对称,电导率越高。
5. 结论
我们采用密度泛函理论研究了立方、斜方六面体、正交和单斜四种结构La0.9Sr0.1Ga0.8Mg0.2O3−δ电解质的氧空位形成能和迁移能。总结如下:
1) 空位形成能和迁移能:计算结果显示立方结构LSGM与斜方六面体、正交和单斜结构相比,具有最小的空位形成能和迁移能,而另外三种结构的空位形成能和迁移能相似且都比较大。
2) 空位迁移能的变化规律:计算的平均空位迁移能按如下顺序增加:
,呈现一个明显的变化趋势:晶格越扭曲(对称性越低),迁移能越高。在正交和单斜结构的LSGM中,两个近邻O2位之间的迁移能要明显低于O1和O2位之间的迁移能,这表明在正交和单斜结构的LSGM中与O1-O2路径相比,被解离后的氧空位更容易沿O2-O2路径发生迁移。
3) 最佳晶体结构:与其他三种结构相比,立方结构的LSGM具有最低的空位形成能和迁移能,因此在四种结构中立方结构LSGM具有最好的氧离子电导。氧离子电导随对称性的变化趋势为:晶格越接近完美立方对称,氧离子电导就越高。
我们计算并比较了立方、斜方六面体、正交和单斜四种结构La0.9Sr0.1Ga0.8Mg0.2O3−δ电解质的氧空位形成能和迁移能,探讨了氧离子电导随对称性的变化趋势:晶格越接近完美立方对称(对称性越高),氧离子电导就越高,此趋势与文献报道其他钙钛矿材料一致,并且预言了立方结构LSGM在四种结构中具有最好的氧离子电导。
基金项目
本项目由2017河南省科技计划项目(No.172102210115);2018河南省科技计划项目(No.182102210140);2018河南省重点研发与推广专项(No.182102210564);河南省高校科技创新人才计划(No.18HASTIT030);河南省大学生创新训练计划项目(201912949007);郑州师范学院科技创新团队支持计划;郑州师范学院大学生创新性实验计划(DCZ2018020 and DCZ2019028)提供经费支持。
参考文献