1. 引言
近年来,六方氮化硼(h-BN)作为一种新型二维材料,其特殊的机械性能及电子性质引起了人们的关注 [1] [2] [3]。单层的h-BN结构与石墨烯类似,B、N原子交替排列形成正六边形。h-BN的热稳定性、绝缘性、宽带隙等特点使它在材料领域有许多潜在的应用。特别是它的高比表面积,使其成为气敏传感器的理想材料之一。目前,已有许多研究报道了掺杂h-BN的气敏特性 [4] [5] [6] [7] [8]。借助于密度泛函理论(DFT) Samadizadeh [4] 等研究了掺杂Al的氮化硼对N2O气体分子的吸附;Zhang等 [5] 利用第一性原理的研究表明CO在缺陷位及Al掺杂的氮化硼纳米片上,可形成稳定的化学作用。因此h-BN在电子器件及气体传感领域有广阔的应用前景。
甲醛(H2CO)是一种常见的室内污染气体,能引发包括白血病在内的多种疾病,严重危害人们的健康。目前已有文献 [9] [10] [11] [12] [13] 报道氮化硼应用于甲醛的吸附。例如,Ye的实验表明h-BN材料对H2CO表现出超高的吸附性能 [9]。Noorizadeh [10] 的理论计算表明H2CO在氮化硼完美表面吸附作用很小,但是在Al掺杂表面形成较强的吸附。Cuba-Supanta等 [11] 计算了H2CO在氢化的h-BN的吸附,计算表明H2CO容易吸附在B端裸露的表面。实际的氮化硼纳米材料可能存在结构上的缺陷,例如空位、边位、吸附原子等,这些缺陷位点对纳米材料的化学性质有显著的影响 [14] [15] [16] [17] [18]。因此本文考察了H2CO在完整的h-BN表面及各种缺陷位的吸附情况,旨在探究H2CO在h-BN吸附的可能性,为设计氮化硼基甲醛检测及消除的新型分子器件提供理论依据。
2. 计算模型与计算方法
选用B27N27簇模型模拟h-BN单层,边缘的原子全部用H原子饱和,如图1(a)所示。在此模型的基础上,分别去掉一个中心N或B原子,可得到N及B空位的模型,分别为图1(b)~(c)。图1(d)~(f)分别用于模拟h-BN的椅式边位、N边位及B边位。
选用色散矫正的密度泛函方法(B3LYP + D3),采用6-311G(d, p)基组进行计算。对所有的原子坐标进行全优化,并结合频率确认该结构没有虚频。计算采用Gaussian16程序包完成 [19]。
吸附能用公式(1)计算:
(1)
其中,E(H2CO_h-BN)代表甲醛吸附在氮化硼的总能量,E(H2CO)代表甲醛分子的总能量,E(h-BN)代表六方氮化硼的总能量。由此公式定义的吸附能负值表示热力学稳定的体系。
3. 计算结果与讨论
3.1. 计算模型的优化结构
图1(a)为完整的h-BN单层结构,计算所得B-N键长为1.448 Å,与实验值1.44 Å [17] 接近。图1(b)、(c)分别为N、B空缺位结构。两个空缺位构型仍基本保持平面构型,并没有发生重构。在N空位构型中,三个相邻的B-B键长分别为2.273 Å、2.273 Å及2.291 Å。在B空位构型中,三个相邻的N-N键长分别为2.739 Å 2.739 Å及2.833 Å,与文献报道较为一致 [18]。图1(d)~(f)为椅式边位、N边位及B边位。从优化构型来看,椅式边位稍有变形:B-N键长为1.295 Å,与完整表面的B-N键长相比,缩短了0.153 Å,这可能是由于边位的B原子上有悬空键,更易与附近的N原子成键。另外值得注意的是,边位上的B-N键不再保持水平,其中的N原子相对于B原子略有上移。图1(e)为N边位的优化构型,边位的B-N键长为1.422 Å,与完整表面的B-N键相差不大。图1(f)为B边位的优化构型,相邻的两个B-N键长分别为1.371 Å及1.481 Å。
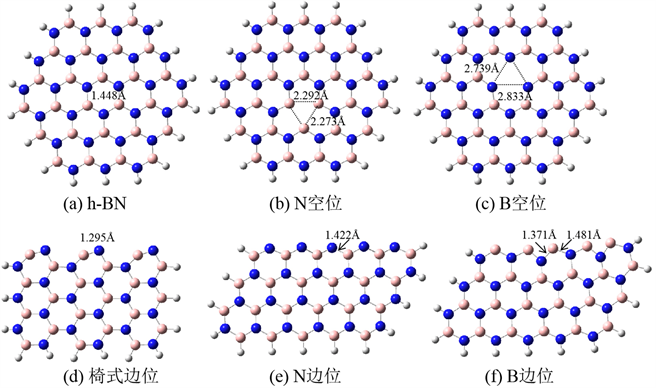
Figure 1. The optimized structures of the computational models
图1. 计算模型的优化结构
3.2. 甲醛在完美氮化硼表面及边位氮化硼上的吸附
图2给出了甲醛(H2CO)在完美及边位h-BN的吸附构型,表1给出了相应的吸附能。图2(a)为甲醛在完美h-BN表面的吸附结构。甲醛分子以垂直方式吸附于BN表面,吸附高度为2.939 Å,吸附能仅为3.4 kcal/mol,是较弱的物理吸附。
图2(b)为甲醛在N边位的吸附构型。甲醛分子中的羰基吸附在相邻的两个边位N上,形成五元环的吸附构型,并且形成的C-N及O-N吸附距离较短,分别为1.461、1.431 Å。吸附后甲醛分子的构型变化较大,C=O键长由气相的1.207 Å拉伸到1.457 Å。说明羰基上的双键已被破坏,原来的平面构型转变为三角锥形,因此吸附后C原子的杂化类型由原来的sp2转变为sp3杂化。从吸附能来看,甲醛吸附后放热69.6 kcal/mol,是强的化学吸附。因此N边位是较好的吸附位点。
图2(c)为甲醛在B边位的吸附构型。与N边位的吸附构型类似,甲醛在B边位也形成五元环的吸附构型,形成的C-B及O-B吸附距离分别为1.608、1.370 Å。其中B-O键长接近HBO3中的B-O键长1.361 Å [20],因此可认为形成了较强的B-O键。吸附后甲醛中的羰基键长为1.474 Å,说明原来的双键已经被破坏,甲醛不再是平面构型。计算得到的吸附能为−131.5 kcal/mol,是非常强的化学吸附。因此B边位对甲醛的吸附能力比N边位更强。这可能是B边位容易形成稳定的B-O键导致的。
图2(d~f)为甲醛在椅式边位的吸附构型。其中,(d)是甲醛以O原子吸附在B位,形成的B-O距离为1.588 Å,甲醛中的C=O键长为1.233 Å,与气相相比相差不大,甲醛仍保持平面构型,可以推测此吸附的吸附强度不大。计算的吸附能为−17.9 kcal/mol,与上述边位的吸附能相比较弱,但仍为化学吸附。(e)是甲醛以C=O基吸附在两个相邻的BN结构单元的B和N原子上,形成六元环的结构。甲醛分子中的C=O键拉伸为1.358 Å,且吸附后变为非平面型,可以推测是较强的化学吸附,吸附能为−34.7 kcal/mol也印证了这一点。(f)是甲醛分子以C=O基吸附在BN的一个六边形结构单元上,形成四元环的结构。C-N及O-B吸附距离分别为1.459、1.399 Å,甲醛中的C=O键长伸长到1.456 Å,H2CO不再保持平面构型,说明吸附作用非常强烈,导致甲醛中的双键被破坏。吸附能为−55.5 kcal/mol,是较强的化学吸附。
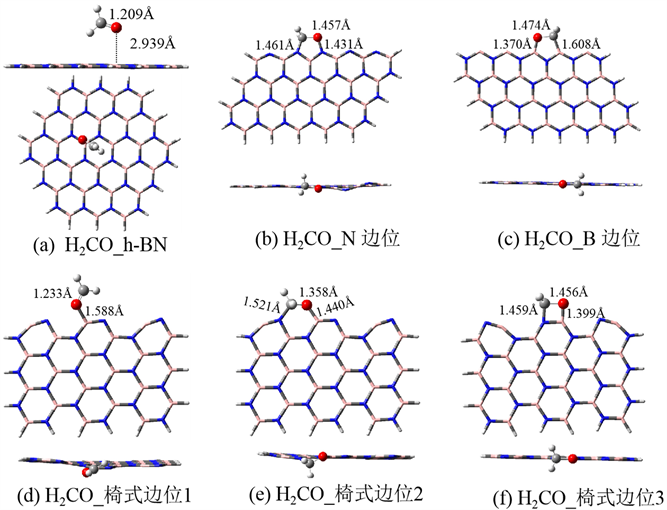
Figure 2. The adsorption structures of formaldehyde on the perfect and edge sites of h-BN
图2. 甲醛(H2CO)在h-BN完整表面及边位的吸附构型
3.3. 甲醛在h-BN空缺位的吸附
图3为甲醛在氮化硼空缺位的吸附构型。图3(a)为甲醛在B空位的吸附结构,甲醛近似平行地吸附在B空位,但是吸附后甲醛的结构几乎没有变化,吸附高度为2.869 Å,吸附能为−4.8 kcal/mol,是弱的物理吸附。与B空位的吸附完全不同,甲醛在N空位的吸附构型如图3(b)所示。甲醛以C-O端吸附在空缺位的两个相邻的B原子上,吸附后甲醛的C=O键长削弱,拉伸到1.437 Å;形成了一个新的B-O键,键长为1.373 Å。因此,甲醛在N空位的吸附很强,吸附能为−75.6 kcal/mol。甲醛在两个空缺位的吸附完全不同,这可能是由于B及N缺位的电子结构完全不同引起的 [21]。另外,N空位容易形成B-O键,导致吸附更易进行。
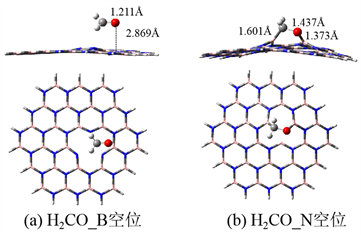
Figure 3. The adsorption structures of formaldehyde on the vacancy of h-BN
图3. 甲醛(H2CO)在h-BN空位的吸附构型
计算表明,甲醛在h-BN的完整表面的吸附较弱,但是h-BN可能存在丰富的边位不饱和位及点缺陷位,使得H2CO容易在这些位置被捕获,形成稳定的化学吸附。在本文讨论的缺陷位中,B边位对甲醛的吸附能最大,其次为N空位,N边位,及椅式边位。虽然椅式边位的吸附相对较弱,但是,椅式边位可以提供丰富的吸附位点,使得H2CO容易在此位置吸附。从吸附构型来看化学吸附会导致H2CO的C=O削弱,C的杂化由sp2转变为sp3,吸附后H2CO不再是平面构型。吸附过程中一旦形成B-O键,会导致能量下降,体系更为稳定,因此是形成化学吸附的主要驱动力。
从表1可以看出,稳定的吸附体系均伴随明显的电子转移。比如当甲醛吸附在B边位、N空位时,甲醛分子明显带负电,说明电荷由h-BN体系向吸附物种转移,导致较强的吸附作用。当甲醛吸附在N边位、椅式位1时,甲醛分子带正电,说明电荷由甲醛向氮化硼体系转移。因此不管电荷转移的方向如何,只要发生明显的电荷转移,将导致较强的化学吸附。

Table 1. The adsorption energy, adsorption distance and charges for H2CO on h-BN
表1. H2CO在h-BN的吸附能、吸附高度及电荷
4. 结论
本文利用密度泛函B3LYP方法计算了甲醛在h-BN表面及缺陷位的吸附构型及吸附能。计算表明,甲醛在完整表面的吸附能力很弱,但是在不饱和位及缺陷位吸附较强,能形成有效的化学吸附。按照吸附能大小,甲醛在缺陷位的吸附由强到弱依次为B边位 > N空位 > N边位 > 椅式边位。因此有望设计以h-BN为基底的甲醛检测或消除材料。
致谢
感谢江苏大学高性能计算平台。
基金项目
黔西南州科技项目(2019-2-51);贵州省普通高等学校青年科技人才成长项目黔教合(KY字[2019]220)。