摘要: 目的:探讨1例KMT2A基因新发突变导致的Wiedemann-Steiner综合征(WDSTS)合并矮身材患儿的临床特点和重组人生长激素(rhGH)治疗效果。方法:收集1例WDSTS患儿的临床资料,进行全外显子基因测序,应用rhGH治疗,并进行文献复习。结果:患儿女,6岁7个月,因“生长迟缓6年”就诊,身高109.0 cm (−2.01 SD),头围47.0 cm (< −2.0 SD),浓眉,眼距宽,眼睑下垂,宽鼻梁,牙列不整齐,牙齿间距宽,指趾短,颈背部、前臂、下肢多毛,基因检测显示患儿KMT2A基因第16号外显子存在新发杂合突变,变异位点c.5005-2A>G,患儿父母无此变异。给予rhGH治疗3月,身高增长3.0 cm,治疗过程中未出现不良反应。结论:WDSTS合并矮身材患儿短期应用rhGH治疗有效,远期需要监测其安全性。
Abstract:
Objective: To investigate the clinical characteristics and therapeutic effect of recombinant human growth hormone (rhGH) in a case of Wiedemann-Steiner syndrome (WDSTS) with short stature caused by a novel mutation of KMT2A gene. Methods: The clinical data of a child with WDSTS were collected, the whole exon gene was sequenced, rhGH was used, and the literature was reviewed. Results: The child aged 6 years and 7 months whose chief complaint was growth retardation for 6 years. Her height was 109.0 cm (−2.01 SD), and head circumference was 47.0 cm (< −2.0 SD). She showed thick eyebrow, wide distance between eyes, ptosis, wide nasal bridge, irregular dentition, wide distance between teeth, short fingers and toes, hairy neck, back, forearm and lower limbs. Gene testing showed that there was a new heterozygous mutation in exon 16 of KMT2A gene, mutation site is c.5005-2A>G. There was no variation in this site in her parents. After rhGH treatment for 3 months, her height increased by 3.0 cm, and there was no adverse reaction during the treatment. Conclusion: Short-term rhGH therapy is effective in children with WDSTS and short stature, and long-term safety monitoring is needed.
1. 引言
Wiedemann-Steiner综合征(WDSTS, OMIM#605130)是一种罕见的常染色体显性遗传疾病,在1989年最早由Wiedemann等人报道 [1],在2000年由Steiner等人首次描述 [2]。2012年,Jones等人使用全外显子组测序发现WDSTS是由组蛋白赖氨酸N-甲基转移酶2 (KMT2)基因杂合性缺失突变引起 [3]。WDSTS临床特征为身材矮小、肘部多毛、发育迟缓、轻度至中度智力低下和特殊面容(长睫毛、浓眉、眼睑下垂、睑裂窄而长、鼻梁宽、人中长、上唇薄等)。本研究我们报道1例WDSTS患儿的临床及基因特点,观察重组人生长激素(rhGH)治疗效果,为临床诊断和治疗提供依据。
2. 病例介绍
患儿女,6岁7个月,因“生长迟缓6年”于2020年12月就诊于本院。患儿系G1P1,母孕期正常,因其母亲有“妊高症”足月剖宫产分娩,出生体重2.3 kg,身长45 cm,生后无窒息抢救史。患儿自生后6个月较正常同龄儿童生长迟缓,年生长速率不详,1岁会叫“爸爸”“妈妈”,2岁会独走,记忆力尚可,计算能力差。父母身体健康,否认近亲结婚,否认家族中遗传病史。父亲身高175 cm,母亲身高165 cm,遗传靶身高163.5 cm。
体格检查:一般情况可,H 109.0 cm (−2.01 SD),Wt 19.5 kg (−0.8 SD),头围47.0 cm (< −2.0 SD)。浓眉,眼距宽,眼睑下垂,宽鼻梁,牙列不整齐,牙齿间距宽,指趾短,颈背部、前臂、下肢多毛,心、肺、腹查体无明显异常,神经系统查体无异常,脊柱无侧弯,四肢脊柱无畸形,双乳B1期,女性外阴,外阴幼稚,阴毛PH1期,无颈短颈蹼,无肘外翻(图1)。
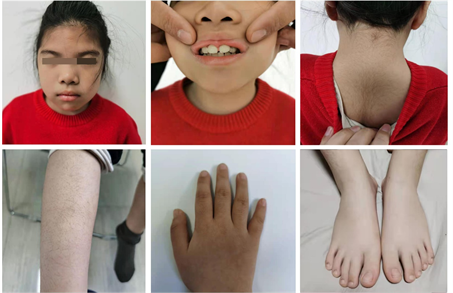 浓眉、眼距宽、眼睑下垂、宽鼻梁;牙列不整齐、牙齿间距宽;颈背部、前臂、下肢多毛;指趾短。
浓眉、眼距宽、眼睑下垂、宽鼻梁;牙列不整齐、牙齿间距宽;颈背部、前臂、下肢多毛;指趾短。
Figure 1. Clinical characteristics of WDSTS child
图1. WDSTS患儿临床特征
辅助检查:采用精氨酸联合可乐定两种药物进行生长激素激发试验,结果提示生长激素(GH)峰值为4.05 ng/ml;胰岛素样生长因子1 (IGF-1) 56 ng/ml;游离三碘甲状腺素原氨酸(FT3) 7.27 pmol/l、游离甲状腺素(FT4) 14.43 pmol/l、促甲状腺激素(TSH) 2.57 uIU/ml;促肾上腺皮质激素(ACTH) 11.30 pg/ml、皮质醇(COR) 7.5 ug/dl;促卵泡激素(FSH) 0.73 mIU/ml、促黄体生成素(LH) 0.16 mIU/ml、雌二醇(E2) 17.82 pg/ml、睾酮(T) 0.09 ng/ml、孕酮(P) 0.24 ng/ml、泌乳素(PRL) 11.6 ng/ml;血常规、尿常规、肝肾功能、电解质、血糖、甲胎蛋白(AFP)、人绒毛膜促性腺激素(HCG)均无异常。外周血染色体核型46,XX;骨龄8.0岁(G-P法),腹部彩超、泌尿系彩超和盆腔B超、脊柱X线及垂体核磁共振均正常,韦氏儿童智力量表测试总分68分。
基因检测:为进一步明确诊断,经患儿父母知情同意后行全外显子基因检测。结果显示(图2):患儿KMT2A基因第16号外显子存在杂合突变c.5005-2A>G,导致氨基酸发生剪接突变(splicing)。一代测序验证结果提示父母均未检出该突变,患儿突变为自发突变。查阅PubMed文献数据库、人类基因突变数据库(HGMD)和疾病相关基因变异数据库(ClinVar),国内外均未有该变异位点的相关报道。根据美国医学遗传学和基因组学协会(ACMG)变异分析指南,该变异判定为致病性变异:证据如下:(1) 该变异为剪接突变,可能导致基因功能丧失(PVS1);(2) 新发变异,患儿父母该位点无变异(PS2);(3) ClinVar数据库对该位点的致病性分析为致病性(PS4);(4) 在正常人群数据库中未有该变异报道,频率为0 (PM2)。
诊断和治疗:该患儿明确诊断为WDSTS。与家长充分沟通,并签署知情同意书后,给予患儿每晚睡前皮下注射rhGH (34 μg/kg/d)治疗连续3月,身高增长3.0 cm,3个月后IGF-1、血糖、甲状腺功能均在正常范围,治疗过程中未出现不良反应,目前仍在继续rhGH治疗中。
3. 讨论
自2012年Jones等人 [3] 首次报道WDSTS患者KMT2A基因突变以来,目前已有70多例报道 [4]。KMT2A基因位于染色体11q23上,由37个外显子组成,跨度超过100 kb。KMT2A基因功能缺失(LOF)突变与WDSTS相关,包括无义突变33%,缺失突变26%,错义突变16%,剪接突变11%,重复突变9%,插入突变5%,以上突变均为致病性或可能致病性 [5]。本例患儿为剪接突变,根据ACMG指南为致病性变异。
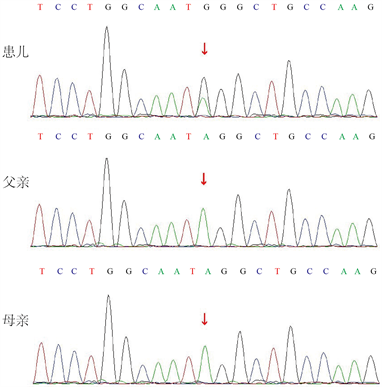 患儿KMT2A基因存在杂合突变c.5005-2A>G,导致氨基酸发生剪接突变(splicing)。父母均未检出该突变。
患儿KMT2A基因存在杂合突变c.5005-2A>G,导致氨基酸发生剪接突变(splicing)。父母均未检出该突变。
Figure 2. Sanger sequencing of KMT2A gene in the child and her parents
图2. 患儿及其父母KMT2A基因Sanger测序图
KMT2A基因主要编码3969个氨基酸的蛋白质。KMT2A为组蛋白H3赖氨酸4位残基(H3K4)甲基转移酶,可以催化组蛋白特定位点加上修饰基团的酶,H3K4甲基化是一种进化上保守的组蛋白修饰。组蛋白与DNA紧密结合,组蛋白修饰在基因表达调控、DNA复制以及DNA损伤修复过程中发挥着重要的作用。KMT2A即是通过与在不同基因活性启动子区开放的染色质结构结合来正向调控许多靶基因的表达,比如多个HOX和Wnt相关基因和其他参与胚胎发育的基因 [6] [7]。因此,WDSTS患者的表型复杂,涉及多个系统,临床症状存在高度异质性。
WDSTS临床表现多样,常见的主要特征包括智力低下、身材矮小、肘部多毛、特殊面容(长睫毛、眼睑下垂、浓眉、眼裂窄、眼距宽、宽鼻梁)。文献报道常见临床表现所占比例分别为:智力低下(98.6%)、身材矮小(70.4%)、长睫毛(67.6%)、眼睑下垂(64.8%)、浓眉(64.8%)、眼裂窄(64.8%)、眼距宽(54.9%)、肘部多毛(52.1%)、背部多毛(52.1%)、喂养困难(49.3%)、宽鼻梁(46.5%)、肌张力低下(43.7%) [4]。而我国较常见的临床表现为长睫毛(94.0%)、智力低下(93.0%)、语言发育落后(86.0%)、眼裂下垂(81.0%)、生长迟缓(75.0%)、背部多毛(75.0%)、宽眼距(75.0%)、骨龄延迟(70.0%)、上睑下垂(63.0%)、宽鼻梁(63.0%)、运动发育落后(63.0%)、下肢多毛(50.0%)、长人中(56.0%)、薄上唇(50.0%)、短指(50.0%)、小头(50.0%) [8]。该患儿临床表现为小头,浓眉,眼距宽,眼睑下垂,宽鼻梁,牙列不整齐,牙齿间距宽,指趾短,颈背部、前臂、下肢多毛,与文献报道一致。该患儿骨龄较实际年龄提前,在Li [8] 等人报道中显示骨龄提前约占20%,骨龄落后占70%,具体机制尚不明确。
身材矮小是WDSTS的一个常见临床表现,生长激素缺乏已经作为WDSTS表型谱的一部分。Baer等人报道50%的WDSTS患者存在生长激素缺乏,但没有描述诊断生长激素缺乏的具体方法,不能排除是否假阳性 [9]。而上官华坤等人报道WDSTS患者合并生长激素缺乏占8.5% [4]。身材矮小的具体机制尚未阐明,KMT2A基因突变可能是其根本原因。George等人报道1例WDSTS患儿合并垂体前叶发育不良伴垂体后叶异位,表明KMT2A基因可能在垂体发育中起关键作用 [10]。另外,组蛋白赖氨酸甲基转移酶(KMT)突变引起的其他综合征也伴有生长障碍,比如KMT2D基因突变引起的Kabuki综合征等 [11] [12]。
文献报道显示短期和长期采用rhGH治疗WDSTS合并矮身材有效:rhGH治疗6月,身高改善明显,肝功能、甲状腺功能、血糖水平正常 [13];rhGH (50 μg/kg/d)治疗12月,身高增长10 cm [14];rhGH (50 μg/kg/d)连续14月,身高增长12 cm [8];rhGH (25~30 μg/kg/d)治疗WDSTS长达6年,疗效显著,身高增长13.3 cm/年,IGF-1和IGFBP-3维持在正常范围,无明显不良反应,WDSTS相关表型特征也无明显改变 [10]。rhGH联合促性腺激素释放激素抑制剂治疗WDSTS合并矮身材以及青春期发育提前患儿,2.5年身高增长20.5 cm,期间未出现药物不良反应 [4]。本例患儿应用rhGH (34 μg/kg/d)治疗3月,身高增长3.0 cm,IGF-1、血糖、甲状腺功能在正常范围,治疗过程中未出现不良反应。本文局限性是该患儿治疗时间较短。因此,即使rhGH治疗WDSTS矮身材短期效果明显,治疗期间临床安全性仍需要长期关注。
4. 结论
综上所述,对于身材矮小的WDSTS患者,需要评估垂体功能,进行生长激素刺激试验和垂体核磁共振检查,WDSTS合并矮身材的患者可以考虑rhGH治疗,临床需要长期监测其安全性。
声明
患儿监护人知情并签署书面知情同意书。
基金项目
临沂市科技发展计划项目(202020003)。
NOTES
*通讯作者。