1. 引言
挥发性有机化合物(VOCs)作为全球型大气污染物是造成城市灰霾、光化学烟雾、大气毒性等复合型大气污染现象的主要推手之一,给人们的日常生活、健康带来严重影响 [1] [2] [3] [4]。催化氧化方法因净化率高、耗能低等优点被认为是净化VOCs最有前景的技术之一 [5] [6]。催化氧化技术的核心是催化剂。在众多催化剂中,铈钴复合氧化物已成为人们研究VOCs净化的热点材料 [7]。氧化铈具有良好的表面碱性、高氧流动性及供氧能力,适当在氧化钴中加入氧化铈可有效改善催化剂的催化性能,提高热稳定性 [8] [9]。但传统的合成方法常需要多步独立且操作较为复杂耗时,难以满足工业规模的催化剂用量需求。同时,异质原子复合如何影响催化剂的晶体结构也有待明确。本文通过简单的一步固相法制备了不同Ce/Co比例的固溶体催化剂,探讨了不同配比铈钴复合对催化剂的结构及可还原性能的影响,考察了水蒸气含量、反应时间及温度等因素对催化剂催化氧化活性的影响。
2. 实验部分
2.1. 实验药品
六水合硝酸铈(Ce(NO3)3∙6H2O,分析纯,艾览(上海)化工科技有限公司),六水合硝酸钴(Co(NO3)2∙6H2O,分析纯,国药集团化学试剂有限公司),柠檬酸(C6H10O8,分析纯,国药集团化学试剂有限公司),甲苯(C7H8,分析纯,西陇科学股份有限公司)。
2.2. 催化剂制备方法
无需添加水等溶剂,利用柠檬酸的自聚特性直接将固体原料混合加热制备五种Ce/Co固溶体催化剂。其中5CoOx-CeOy固溶体的具体制备方法如下:按照摩尔比5:1:9分别称取Co(NO3)2∙6H2O、Ce(NO3)3∙6H2O及柠檬酸进行重复混合。将混合物置于马弗炉中以5℃/min升温速率至450℃并维持2 h,得到5CoOx-CeOy。其他摩尔比Co-Ce样品及CoOx、CeOy制备方法与上述方法类似。
2.3. 催化剂表征
采用X-ray Diffraction (岛津,XRD-7000)分析催化剂的晶体结构,测试条件为:Cu靶Kα辐射源(λ = 0.15406 Å),扫描速度2θ = 2˚/min,扫描范围为10~70˚,电压40 kV。采用AutoChem化学吸附仪(美国Micrometitics)检测催化剂的氢气程序升温还原能力(H2-TPR):0.05 g催化剂装入U型石英反应管中,先在300℃下以50 ml∙min−1惰性气体Ar气流进行吹扫处理1 h,待炉温降至室温时,将进气氛围改为50 ml∙min−1 5 vol%H2/Ar的混合气,系统稳定后,以10℃∙min−1的升温速率升至1000℃,H2消耗量通过热导检测器(TCD)检测。
2.4. 催化剂的催化性能测试
将催化剂装入固定床催化反应管中部并固定于管式炉中,设定管式炉升温至预定值。直接以空气为平衡气体,通过质量流量计控制系统总气体流量为100 ml∙min−1,设置空速为WHSV = 30000 mL∙g−1∙h−1,调节进口甲苯浓度约为1000 ppm。VOC的转化率(
)、CO2收率(
)以及VOC总氧化和CO2产生的表观活化能
和
采用下列公式进行计算:
(1-1)
(1-2)
(1-3)
(1-4)
式中
、
则分别为进出口的VOC浓度,单位为mol∙m−3;
和
表示进出口的CO2浓度,单位为mol∙m−3;
代表VOC的总氧化速率,
则为转化为CO2部分的VOC氧化速率(mol∙m−3∙s−1);n和m分别为VOC氧化和CO2收率(矿化部分的VOC)的表观反应常数;R为气体常数,T为热力学温度(K),A为指前因子。
3. 结果与讨论
3.1. 催化剂结构分析
从图1中可以看出,CoOx的所有XRD衍射峰与 的标准卡片PDF#43-1003相对应,CeOy的所有衍射峰则与CeO2的标准卡片PDF#43-1002相对应,这表明当前所制备的CoOx和CeOy样品分为Co3O4和CeO2。而当将CoOx和CeOy复合后可以看出,5CoOx-CeOy固溶体催化剂中所有衍射峰峰强均不明显,且对应于CeO2和Co3O4特征峰的半峰宽显著增大,表明催化剂晶体结构发生松弛畸变,这有利于催化剂表面氧物种发生迁移转化而产生更多的表面活性氧,促进催化剂对甲苯的催化氧化。
图2为CoOx、CeOy和5CoOx-CeOy的H2-TPR曲线。对于CoOx催化剂,337℃处的还原峰对应于Co3+到Co2+的还原,位于395℃还原峰归属于Co2+到Co0的还原。而对于CeOy催化剂,其位于508和727℃特征峰则分别对应于Ce4+到Ce3+的转化和Ce3+到Ce2+的还原 [10]。当将两者复合后可发现,获得的5CoOx-CeOy所有特征峰均向低温偏移,尤其是对应于Co4+→Co3+和Co3+→Co2+的还原峰,其分别向低温移动了55和51℃。这表明复合催化剂晶格氧的可迁移性变得更好,也间接表明5CoOx-CeOy比CoOx和CeOy样品可对甲苯产生更高得催化氧化作用。
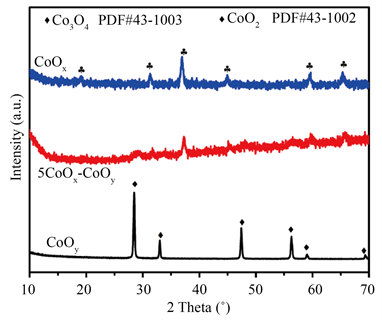
Figure 1. XRD patterns of CoOx、CeOy和5CoOx-CeOy
图1. CoOx、CeOy和5CoOx-CeOy的XRD图

Figure 2. H2-TPR of CoOx、CeOy和5CoOx-CeOy
图2. CoOx、CeOy和5CoOx-CeOy的H2-TPR图
3.2. 不同Ce/Co比对催化活性的影响
从图3(a)中可以看出各个催化剂催化氧化甲苯的去除率均随着温度的升高而增加,且在反应温度低于220˚C时,CeOy的催化氧化活性明显高于CoOx,但温度进一步提升后,CoOx催化氧化活性出现突增并反超CeOy。该现象表明CoOx低温催化氧化活性不足,但当反应温度越过临界点时,其表面大量的反应位活性点便被激活。另外,CeOy尽管具有比CoOx要好的低温催化氧化活性,但其活性增势随温度变化却较为缓慢,显然不利于进一步的工业化应用。当掺入少量铈时,催化剂(6CoOx-CeOy)的低温催化氧化甲苯活性便得到了大幅增强;但过量的铈掺杂减少了复合催化剂中钴的含量(3CoOx-CeOy),不利于复合催化剂中出现更多的反应活性位点。故随着铈含量不断增加,催化剂低温催化活性呈现先增大后减小的趋势,不同铈含量掺杂催化剂在220℃前呈现的催化氧化效果变化趋势如下:5CoOx-CeOy > 4CoOx-CeOy > 6CoOx-CeOy > CeOy > 3CoOx-CeOy > CoOx,其中催化剂5CoOx-CeOy获得的低温甲苯氧化效率最好,说明钴与铈的比例为5:1时两种氧化物复合后,催化剂表面可发生低温迁移的活性氧数量最多,有利于甲苯的低温催化氧化。
 (a) (b)
(a) (b)
Figure 3. (a) Total conversion of toluene oxidation and (b) carbon dioxide yield over different cerium content catalysts
图3. 不同铈含量催化剂作用下的甲苯氧化总转化率(a)和(b)二氧化碳收率
图3(b)为不同铈含量催化剂催化氧化甲苯获得的CO2收率曲线,6CoOx-CeOy、5CoOx-CeOy、4CoOx-CeOy、3CoOx-CeOy变化趋势与甲苯的催化氧化效率曲线相一致,且5CoOx-CeOy催化氧化甲苯所得CO2高低温下均最佳。但CeOy和3CoOx-CeOy的CO2收率曲线与其总催化氧化甲苯效率相差较大。前期研究表明,在VOCs的催化氧化过程中,由于晶格氧(O2−)核外已经带有两个电子趋于饱和状态,其并不具有催化矿化能力,而吸附氧(O−)核外电子处于缺电子状态,所以VOCs矿化为CO2本质上取决于可用的吸附氧含量。因此,氧化铈虽然具有好的储能氧能力,但其主要是以晶格氧形式存在,并没有把自身存储的晶格氧大量转化为可进攻矿化甲苯的表面吸附氧,这也导致了其在等当量的使用下没有较好的矿化效率。相反,CoOx在反应温度高于220℃后,其CO2收率迅速提升,这表明CoOx表面的晶格氧、氧空位、吸附氧三者之间的协同转化能力远高于CeOy。但从图3(b)中也可发现在反应温度低于220℃时,纯CoOx的低温矿化能力很差,这表明其表面的晶格在该温度范围内很难被激活发生“晶格氧→吸附氧”之间的转化。所以将CoOx与CeOy复合,可有效将CeOy丰富晶格氧的易迁移性和CoOx快速转移晶格氧为吸附氧化的特性相结合,提升固溶体催化剂的低温矿化能力。
为了进一步考察不同催化剂催化氧化甲苯活性,我们以甲苯总氧化效率达到50%和90%时的反应温度作为比较参数,如表1所示。很明显,每个催化剂的CO2收率温度都要略高于其对甲苯的总氧化转化率,说明该类催化剂在催化氧化过程中存在一定量的副产物。事实上,绝大多数金属氧化物在催化氧化VOCs过程中,如果相应反应温度下激活的表面活性氧数量不足以实现有机物的完全矿化,则会造成部分氧化有机中间体在反应系统中的残留 [11] [12]。
3.3. 催化反应动力学研究
在氧气过量的情况下,VOCs的氧化反应服从一级动力学机制 [13]。根据上文中列出的公式(1~3)和(1~4)可计算出VOCs转化率(
)和CO2产率(
)的表观活化能值,如图4(a),图4(b)所示。甲苯氧化的
和
值依次递增排序为5CoOx-CeOy (93.06和111.62 kJ∙mol−1) < 4CoOx-CeOy (153.16和173.45 kJ∙mol−1) < 6CoOx-CeOy (162.19和181.63 kJ∙mol−1) < 3CoOx-CeOy (197.92和211.88 kJ∙mol−1) < CeOy (221.86和260.03 kJ∙mol−1) < CoOx (270.59和281.49 kJ∙mol−1),可见5CoOx-CeOy具有更好的表面晶格氧到吸附氧的转化能力,使得甲苯在催化剂表面的氧化转化及矿化更容易进行。此外,对比各个催化剂的
值和
值可发现两种间存在明显的差值,即
>
。众所周知,活化能代表了催化剂发生催化反应的难易程度,活化能越低,反应越容易发生。甲苯的总催化氧化仅仅需要甲苯侧链上甲基(
)中的C-H发生断裂即可表现出甲苯数量的减少,而甲苯的矿化反应则需要甲苯中的所有C-H键均发生断裂并被氧化成CO2,显然甲苯发生矿化反应时需要消耗更多的能量 [14]。

Table 1. Comparison of catalytic activities over different catalysts
表1. 不同催化剂的催化活性比较
 (a) (b)
(a) (b)
Figure 4. Kinetic fit curve of total catalytic oxidation of toluene (a) and CO2 yield (b)
图4. (a) 总催化氧化甲苯的动力学曲线和(b)二氧化碳收率的动力学曲线
3.4. 水蒸气对催化反应活性的影响
从图5可以看出,随着水蒸气路流量占比的不断增大,甲苯的总氧化效率几乎不变,而甲苯的矿化率却出现持续的衰减。该现象主要与甲苯氧化与矿化的耗氧情况有关。甲苯发生总氧化反应仅需要发生第一步甲基氧化,其消耗的氧气量很少,当前反应气氛含氧量的下降不足以对其氧化反应产生明显影响。甲苯矿化一般存在如下多步降解过程 [15] [16] [17]:甲苯→苯甲醇→苯甲醛→苯甲酸→·····→二氧化碳。多步反应需要消耗更多的氧,氧气占比的减少显然会影响矿化效果。同时该段范围内转化率不变而CO2收率减少,也说明水蒸气的增加会造成催化反应中间体不能及时被进一步氧化,导致反应尾气中出现副产物(如苯甲醛、苯甲酸等)。
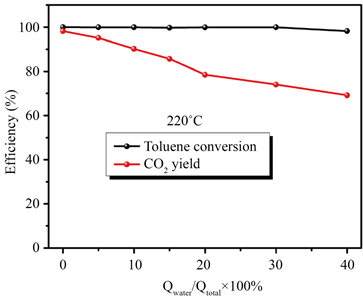
Figure 5. Effect of water vapor on the catalytic activity of 5CoOx-CeOy
图5. 不同水蒸气对5CoOx-CeOy催化剂催化活性的影响
3.5. 空速对催化反应活性的影响
图6为不同空速下5CoOx-CeOy催化氧化甲苯转化率和CO2收率变化曲线。从该图中可以看出,在30000~60000 mL∙h−1∙g−1空速值WHSV范围内,5CoOx-CeOy催化氧化甲苯的转化率和其CO2收率都表现出降低趋势。在反应温度为220℃且氧含量过量状态下,单位时间内催化剂表面产生的活性氧数量几乎维持恒定值,而高空速却增加了单位时间内催化剂表面甲苯含量,增加了催化剂表面活性氧对甲苯分子的氧化负担,导致其甲苯氧化转化和其二氧化碳收率值出现下降趋势。此外,从图中也可发现空速低于30000 mL∙h−1∙g−1时,在220℃的反应温度下,甲苯氧化转化率和二氧化碳收率值几乎都为100%,说明此时催化剂表面产生的活性氧数量对于甲苯矿化已足量。

Figure 6. Effect of WHSV on the catalytic activity of 5CoOx-CeOy
图6. 空速对5CoOx-CeOy催化剂催化活性的影响
4. 结论
本文通过将硝酸铈、硝酸钴及柠檬酸固态原料直接混合,利用柠檬酸的受热自聚特性,直接合成了不同比例的铈钴固溶体催化剂。以甲苯作为VOCs模拟污染物,探讨了钴、铈不同配比对催化剂催化甲苯活性的影响。研究结果表明,CeOy和CoOx的复合增强催化剂表面氧的可迁移性,提升了催化剂的氧化还原能力。其中Co与Ce的摩尔比达到5:1时,复合催化剂可取得最佳的低温催化氧化活性和更低的表观活化能值(Ea(VOC) = 93.06 kJ∙mol−1和Ea(CO2) = 111.62 kJ∙mol−1)。此外,随着水蒸气占比不断增大,甲苯的总氧化效率和甲苯的矿化率却出现持续的衰减现象。本文也为研究掺杂异质原子提升催化剂催化氧化来自大气中的有机污染提供了思路。
基金项目
江苏省高等学校自然科学研究面上项目(22KJB610022),江苏省重点研发计划项目(BE2022767)。
NOTES
*通讯作者。