1. 引言
蛋白质质谱仪是目前鉴定和定量肽段和蛋白的主要工具。数据依赖获取(Data Dependent Acquisition, DDA)和数据不依赖获取(Data Independent Acquisition, DIA)是常用的两种数据采集方式。鸟枪法实验是典型的DDA采集方式 [1] ,质谱仪会先对肽段母离子进行全扫描,获取一级质谱,然后选择一级质谱中强度排名前N的肽段母离子进行碎裂,得到二级质谱。DDA采集方式获取的一级质谱和二级质谱对应关系清晰,但对母离子强度有很高的依赖性,导致实验结果存在随机性,且低丰度肽段的识别性较差。
相比之下,DIA采集方式将母离子质荷比区间划分为多个独立窗口,依次将每个窗口内的所有母离子打碎并记录所有碎片子离子的信息作为二级质谱。DIA采集方式具有实验结果重复性高、数据通量高、灵敏度高等特点,但是由于DIA数据中的碎片子离子信号来自于不同母离子,且混合在同一张二级质谱中,导致DIA质谱数据的分析极为困难。此外,DIA质谱数据的准确定量是生物信息学中的基础问题,科学家在此基础上展开一系列具有生物意义的研究,例如基于SWATH-MS技术(一种DIA质谱技术)对细胞凋亡和坏死性凋亡途径之间串扰的定量理解研究 [2] 和在SWATH-MS质谱数据的基础上进行生物动力学建模 [3] 。对DIA质谱数据的高质量定性定量对这些研究非常重要,因此,开发对DIA质谱数据进行定性定量分析的工具一直是研究热点。
分析DIA质谱数据,常用的研究方法主要分为两种。一种是依赖库分析法,通过DDA实验数据创建文库,将实验输出DIA数据中的谱图与文库中的谱图进行匹配,从而完成肽段定量分析,此种研究方法中的谱图库可以是由大量鸟枪法DDA实验生成外部库,也可以是直接从SWATH-MS数据中的伪质谱文件生成内部库 [4] 。基于此种研究方法的常用工具有OpenSWATH [5] 、SWATHProphet [6] 和Specter [7] 等。
另一种研究方法称为无库分析法,可以省去DDA实验。2015年Tsou C C等人提出的DIA-Umpire [8] 利用肽段母离子的碎裂效率小于100%的特点,计算一级质谱中的肽段母离子和二级质谱中被打碎肽段的相关性,以此找出一级质谱和二级质谱的对应关系,通过这种方式建立伪数据库,实现无库分析。同样基于无库分析法的工具还有Group-DIA [9] 、PIQED [10] 和directDIA (a part of Spectronaut [11] )等。
近几年,基于深度学习的无库分析方法逐渐发展起来。2019年Bernhard Kuster和Mathias Wilhelm提出的Prosit算法,基于RNN模型准确预测母离子的理论谱图和驻留时间,从而获得更加精确的质谱鉴定 [12] 。同年,Tarn等提出的DeepNovo-DIA算法,将深度学习应用于从头测序(de-novo sequencing)法,直接鉴定肽段氨基酸序列 [13] 。2021年,厦门大学教授帅建伟和博士何情祖等人提出算法Ultra-DIA,利用深度变分自动编码器提取离子信号的特征,从而完成对肽段和蛋白进行定性和定量分析 [14] ,第二年该团队基于该算法进行优化并提出以谱图为中心的算法Dear-DIA,结合深度变分自动编码器和三重态损失来学习提取的碎片离子色谱图的特征,然后使用k-means聚类算法将具有相似特征的碎片聚合到同一类中,从而处理DIA质谱数据 [15] 。同年,厦门大学韩家淮院士和俞容山教授团队提出了基于深度学习LSTM的鉴定软件DreamDIA。该软件随机选取部分谱库结果,归一化其保留时间,并对谱库的保留时间进行拟合,从而对剩余肽段的保留时间进行预测。接着,DreamDIA结合LSTM和全连接网络对输入数据进行打分,选择最优匹配结果 [16] 。
在这些研究中,于2020年由Markus Ralser等人开发的DIA-NN是将深度学习用于DIA蛋白质组学数据处理的集成软件包,开启了蛋白质组学的新篇章 [17] 。DIA-NN能够将高通量方法用于可靠,稳健和定量准确的大规模实验。尽管DIA-NN通过算法控制假阳性率,但仍然会输出一定比例的低置信度肽段,这使得生物学研究人员在进行下一步精确研究时,需要人工筛选出高置信度肽段。人工筛选的步骤是先通过可视化软件提取碎片子离子色谱峰组(XICs),然后以六条碎片子离子的色谱峰形相似度为标准筛选出高置信度肽段。目前用于色谱峰可视化的工具有Skyline [18] 、TOPPView [19] 、MSSort-DIAXMBD [20] 和DrawAlignR [21] 。
其中,于2022年李一鸣等人提出的MSSort-DIAXMBD作为OpenSWATH的最后一步,对肽段的 MS/MS数据进行可视化和分类肽段母离子 [20] 。该方法利用OpenSWATH输出报告信息对每个肽段和匹配的碎片子离子组进行色谱曲线重构和可视化,并使用深度卷积神经网络对重构后的信息进行数据挖掘,结合双阈值分割策略,自动识别高置信度肽段和低置信度肽段,本质上是针对OpenSWATH质谱数据分析软件输出肽段的再筛选,缓解人工检查任务负担。
与OpenSWATH相同,DIA-NN的输出报告中普遍包含数万甚至十几万个肽段,如果一个输出报告中包含1000个肽段,检查一个肽段对应的碎片子离子的XICs图片花费30秒,后期的人工检查环节就达到约83个小时,因此人工筛选的过程非常耗时。此外,人工筛选的主观性强,筛选标准不统一,这会导致错误率因人而异。人工检查筛选高置信度肽段如图1所示主要分为两步:利用输出报告的信息(母离子的质荷比和保留时间信息以及质谱仪质荷比窗口信息)从质谱原始数据(母离子保留时间附近的一系列二级质谱信息)提取出每个肽段匹配的碎片子离子组的色谱曲线,存储为图片格式;第二步:科研人员观察色谱图片,以六条子离子色谱曲线相似为标准,删除低置信度肽段,筛选出高置信度肽段。
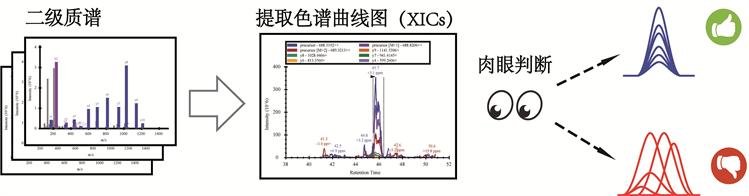
Figure 1. Steps of manual inspection
图1. 人工检查步骤
在本文中,我们提出了一种基于深度学习的算法MSDeepFilter,可以对质谱数据软件DIA-NN报告的肽段数据进行自动且高速的再次分类筛选,从而筛选出高置信度的肽段,为肽段筛选提供了统一标准,替代了繁琐的人工筛选过程。为了测试该模型的分类性能,我们开发了肽段子离子色谱峰可视化工具getXIC,将DIA-NN报告的肽段人工标记成高置信度与低置信度两类,以此创建了包含86,443条肽段的基准测试数据集。我们使用该数据集对MSDeepFilter的模型进行训练和测试。测试结果表明,MSDeepFilter在区别高置信度和低置信度肽段方面表现出色,其可以作为人工筛选DIA-NN输出肽段的替代方法。
2. 建立数据集
我们建立了高质量的基准数据集来优化深度学习模型的参数。基准数据集的原始DIA质谱数据来自不同物种、不同质谱仪生产厂商的质谱仪。我们使用DIA-NN作为主要的定量分析软件,并加入了传统定量工作流OpenSWATH-PyProphet-TRIC (OSPT)的分析结果来增强深度学习模型的泛化能力。
2.1. 数据集来源
表1记录了数据集来源信息,包含数据集名称、质谱仪型号、质谱仪所属公司和所使用的数据分析工作流。按照名称对数据集的基础实验信息进行介绍:
注:第一列为数据集的名字;第二列为采集该数据集原始DIA数据的质谱仪名称;第三列为质谱仪的生产厂商;第四列为处理该数据集的分析软件;其中第二列仪器中的TTOF为质谱仪TripleTOF简称,Lumos为质谱仪Orbitrap Fusion Lumos Tribrid的简称;第四列分析软件中的OSPT workflow为OpenSWATH-PyProphet-TRIC的简称。
数据集Yeast_NN (下载地址:http://www.proteomexchange.org,PXD031160)是由质谱仪TripleTOF 6600 (生产厂商:ABSciex)获取的酵母样品数据。一级质谱质荷比范围为400~1250,包含40个可变窗口;数据集Human_NN是由质谱仪Orbitrap Fusion Lumos Tribrid (生产厂商:Thermo Fischer Scientific)获取的人类样品数据 [22] ,一级质谱质荷比范围为400~1250,包含30个可变窗口;数据集E. coli_NN是由质谱仪TripleTOF 6600获得的大肠杆菌样品 [23] ,一级质谱质荷比范围为400~1250,包含100个可变窗口;数据集HYE124_NN和HYE110_NN是由TripleTOF 5600或TripleTOF 6600质谱仪获得的人类、酵母和大肠杆菌的混合样品数据 [24] ,一级质谱质荷比范围为400~1200,包含32个或64个窗口,窗口大小固定或可变;数据集L929_NN是由TripleTOF 5600质谱仪以SWATH模式获得的小鼠样品数据 [9] ,一级质谱质荷比范围为400~1150,包含100个可变窗口,一式三份重复样品。这6个数据集经过DIA-NN处理且人工贴好标签的肽段总量为42,443,其中高置信度肽段22,306个,低置信的肽段20,137个。
数据集Yeast_OSPT (下载地址:http://www.proteomexchange.org,PXD028735)是由质谱仪TripleTOF 5600获得的酵母样品数据,一级质谱质荷比范围为400~1200,包含64个固定窗口;数据集SGS_OSPT是由质谱仪TripleTOF 5600获得SGS [5] 人类样品数据,一级质谱质荷比范围为400~1200,包含32个固定窗口;数据集Hela_OSPT是由质谱仪Q Exactive HF-X (生产厂商:Thermo Fischer Scientific)获得的HeLa [25] 细胞样品数据,一级质谱质荷比范围为350~1650,包含45个窗口;数据集E. coli_OSPT是由质谱仪TripleTOF6600获得的大肠杆菌样品数据 [23] ,一级质谱质荷比范围为400~1250,包含100个可变窗口;数据集L929_OSPT是由TripleTOF 5600质谱仪以SWATH模式获得的小鼠样品数据 [11] ,一级质谱质荷比范围为400~1150,包含100个可变窗口。数据集BGS_OSPT是由Orbitrap Fusion Lumos质谱仪获得的BGS [26] 小鼠样品数据,一级质谱质荷比范围为350~1650,包含40个窗口;数据集HYE110_OSPT和HYE124_OSPT是由TripleTOF 5600或TripleTOF 6600质谱仪获得的人类、酵母和大肠杆菌的混合样品数据 [24] ,一级质谱质荷比范围为400~1200,包含32或64个窗口,窗口大小固定或可变。这8个数据集经过OpenSWATH-PyProphet-TRIC工作流分析输出结果肽段后,经过人工贴标签的肽段总量为44,316,其中高置信度肽段22,744个,低置信的肽段21,572个。
因此,本文建立数据集总样本数为86,759,其中高置信度肽段45,050个,低置信的肽段41,709个。
2.2. 软件和参数设置
DIA质谱数据分析流程为:首先实验人员制备所需样品,然后通过质谱仪采集DIA数据,得到原始数据,同时在uniprot蛋白质数据库中下载对应物种的蛋白质序列数据库,之后将原始数据和对应物种的蛋白质序列数据库一同输入分析工作流(图2(A))。工作流DIA-NN或OpenSWATH-PyProphet-TRIC对数据进行处理,输出报告中包含识别得到的肽段以及对应的量化结果,最后MSDeepFilter算法筛选得到高置信肽段(图2(B))。
目前,DIA-NN是基于深度学习的DIA质谱数据分析领域使用最广泛的工作流之一。OpenSWATH-PyProphet-TRIC工作流为使用最广泛的传统工作流,结果经过大量实验验证。接下来介绍在数据准备中,两个定量工作流中软件的参数设置。
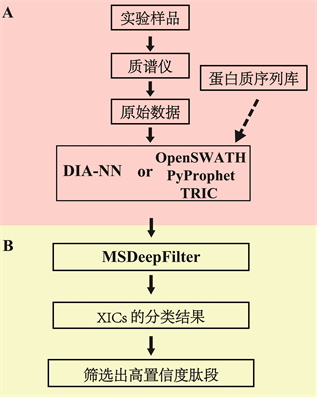
Figure 2. The workflow of DIA-NN & OpenSWATH-PyProphet-TRIC (A) and the workflow of MSDeepFilter (B)
图2. DIA-NN和OpenSWATH-PyProphet-TRIC的工作流程(A)和MSDeepFilter的工作流程(B)
2.2.1. DIA-NN工作流
由于DIA原始文件来源于不同的质谱仪器,导致原始数据文件格式不同,常见的原始格式为.wiff和.raw格式。DIA-NN需要配合MSConvert (V.3.0.19311)和Thermo MS File Reader (3.0 SP3)软件来读取这两种格式的数据文件。我们将DIA数据和蛋白质序列数据库作为DIA-NN (version: 1.7.11)的输入,然后设置一级质谱质荷比范围400~1200,二级质谱质荷比范围100~1800,设置FDR为1%,其他参数为默认值,最后运行DIA-NN获得定量输出报告(图2(A))。
2.2.2. OpenSWATH-PyProphet-TRIC工作流程
首先,我们使用MSConvert (V.3.0.19311)将质谱数据的原始文件转换为mzXML文件,并采用DIA-Umpire生成伪DDA谱图的mgf文件。然后,我们以UniprotKB/Swiss-Prot数据库为参考库,使用TPP (Trans-Proteomic Pipeline, V5.1.0)软件包中的Comet (V2017.01)和X!Tandem (V2013.06.15.1,模式为native和k-score)搜索引擎对伪DDA文件进行数据库搜索,并输出pep.xml格式的搜索结果。下一步,我们使用参数设置为-p0.05 -l7 -PPM -OAdPE -dDECOY的PeptideProphet和参数设置为DECOY = DECOY的iProphet对该搜索结果进行验证和评分。Mayu (V 1.07)用于确定对应于1%肽FDR的iProphet概率。通过1% FDR筛选得到的肽离子被输入到SpectraST中,用于生成定量需要的库文件sptxt。spectrast2spectrast_irt.py脚本(可从http://openswath.org/en/latest/下载)将sptxt文件中肽段的保留时间替换为iRT时间,其中用于保留时间归一化的iRT肽段为内源性肽段。最后,我们使用spectrast2tsv.py脚本将sptxt文件转换为tsv格式,将其转换为TraML格式文件,由OpenSWATH-PyProphet-TRIC工作流定量分析。
2.3. 数据集构成和预处理
我们得到DIA-NN和OpenSWATH-PyProphet-TRIC工作流的输出报告后,通过脚本getXIC.py从DIA原始数据中提取子离子色谱峰组作为MSDeepFilter的模型的输入数据(图2(B))。由于DIA-NN输出的碎片子离子的数量不固定,根据经验(传统人工检查以及常用的数据分析软件计算色谱曲线相关性公式时都使用的强度Top 6的碎片子离子),我们提取强度最高的六个作为子离子峰组成员。通过测试40、55、70、85、90、105、120等不同曲线长度的数据,在比较分类性能指标后,我们最终决定取85个时间点作为时间长度。MSDeepFilter的输入数据为6条长度为85的碎片子离子XICs构成6行85列的矩阵。每条XIC以强度最高点的时间作为中心点,向前取42个时间点的强度数据,向后取42个时间长度的强度数据,然后利用sklearn库中的minmax函数将强度归一化至0到1之间,以消除数量级影响。归一化后的两类样本数据如图3所示。
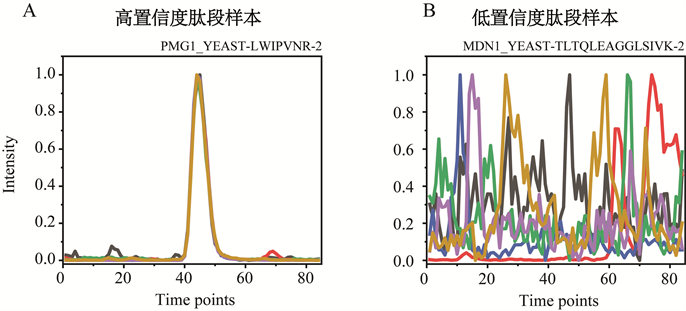
Figure 3. The XICs of High confidence PMG1_YEAST_LWIPVNR and (A) and the XICs of low confidence peptide MDN1_YEAST-TLTQLEAGGLSIVK (B)
图3. 高置信度肽段PMG1_YEAST_LWIPVNR的子离子提取色谱峰组的XICs图(A)和低置信度肽段MDN1_YEAST-TLTQLEAGGLSIVK的XICs图(B)
Minmax归一化函数公式为:
归一化之后对数据进行人工贴标签,数据标签为两类:高置信度肽段和低置信度肽段。贴标签标准为:六条子离子标准化后的色谱峰形状相似则判断为高置信度肽段,反之判断为低置信度肽段。
MSDeepFilter的基准测试数据集包含86,443个肽段,其中高置信度肽段数量为:40,641,低置信度肽段数量为:45,802,将该数据以6:2:2的比例随机分为训练集,交叉验证集和测试集,数量信息如表2所示。
3. 算法
MSDeepFilter的模型原理
为了解决分类问题,我们结合残差网络 [27] 原理、压缩激励模型 [28] 和自注意力机制模块 [29] 原理设计出神经网络结构作为MSDeepFilter的模型。
为了评估MSDeepFilter模型的性能,我们在测试集上应用两种机器学习模型:AdaBoosting [30] 和SVM [31] ,对子离子XICs进行分类,并比较分类性能,这两个模型的参数都是人工调参得到的。
MSDeepFilter的神经网络框架基于残差网络,结合压缩激励模型和自注意力机制设计构成,可以视作三个部分连接组成。模型第一部分包含一个卷积核大小为1 * 1的卷积层。第二部分为残差结构,残差结构包含两条传播路线,残差结构后并行连接压缩激励模块和自注意力机制模块,第三个部分包含一个为神经元个数为16,激活函数为Relu的全连接层,最后的输出层为神经元个数为1,激活函数为Sigmoid的全连接层,最后输出层输出结果(图4)。
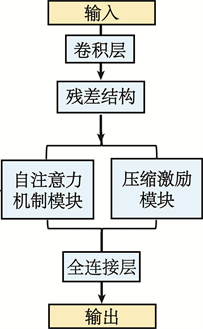
Figure 4. The proposed neural network of MSDeepFilter
图4. MSDeepFilter的模型原理
神经网络输入数据为色谱信息,设计为图像类型结构的三维数据,即(1 × 6 × 85)的三维矩阵,输出为一个实数,大小范围为[0, 1],代表输入肽段为高置信度肽段的预测概率。
MSDeepFilter模型中,卷积层擅长处理多维数组,自动学习给定数据的潜在空间相关性,残差结构中第二条传播路线为跳跃连接的变形,最大程度的保留了数据的原始信息,减轻由于网络结构导致的信息损失带来的模型退化问题。模型中的压缩激励模块通过显式地建模通道之间的相互依赖性,自适应地重新校准通道特征响应,提高模型的性能。(图4中压缩激励模块)。
自注意力机制模块关注数据内部的相关性,使模型聚焦重点信息,有助于提高模型性能(图4中自注意力机制模块)。自注意力机制公式为:
其中的矩阵阵
分别为Q、K、V的权重矩阵,为可训练参数,
为常数,这里我们设置为为通道值,防止softmax输入值过高,导致偏导数趋于0。
我们使用Adam优化器训练MSDeepFilter 模型的神经网络,训练批量大小为256,参数beta1为0.9,参数beta2为0.999,参数epsilon为1e−5,权重衰减为1e−5,学习率为3e−6,训练次数设置为100,损失函数为二元分类的交叉熵损失。
机器学习模型的输入数据维度均为一维,单个样本为大小为6 * 85 = 510的一维数据。
支持向量机(Support Vector Machine, SVM)是一种机器学习方法,可以最大化训练模式和决策边界之间的边距(图5(A))。我们训练了sklearn库里的sklearn.svm.SVC模型,将“kernel”参数设置为“rbf”,“gamma”参数设置为1e−6,参数“C”设置为1e−6,其他参数均设置为默认值。

Figure 5. The model of SVM (A) and the model of AdaBoosting (B)
图5. 支持向量机原理图(A)和AdaBoosting原理图(B)
AdaBoosting属于集成学习中的boosting方法,首先,用初始权重训练得到第一个弱学习分类器,根据弱学习器的学习误差率来更新训练样本的权重,提高使第一个弱分类器学习误差率变高的训练样本的权重,让后续的弱分类器更加重视这些使误差率高的样本。然后,用调整权重之后的训练数据集训练第二个弱分类器,重复训练其余的弱学习器,直到弱分类器数量达到预先设置的值。最后,将所有弱分类器组合得到一个性能更好的分类器(图5(B))。我们训练了sklearn库里的adaboosting模型,将基础弱分类器设置为决策树,决策树参数最大深(max_depth)设置为2,内部节点再划分所需最小样本数(min_samples_split)设置为20,叶节点所需的最小样本数(min_samples_leaf)设置为5;集成算法设置为“SAMME”,基分类器提升(循环)次数(n_estimators)设置为200,学习率(learning_rate)设置为0.8,将其他参数值设置为默认值。
4. 训练测试结果
4.1. MSDeepFilter的模型训练过程
MSDeepFilter的深度学习模型的训练损失函数为结合了Sigmoid激活函数的二元分类的交叉熵损失,其公式为:
其中pred为算法输出的预测值,label是人工标签0或者1。
从图6可看到MSDeepFilter的模型训练次数在80次之后,交叉验证集的损失趋于平稳,最终训练次数为100次,训练结果未出现过拟合或欠拟合问题,说明模型训练成功。
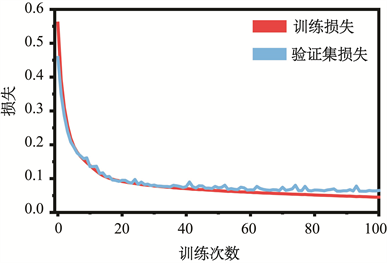
Figure 6. The losses of the proposed networks on the dataset
图6. 模型训练损失随训练次数变化曲线
4.2. 不同模型在数据集上训练测试过程输出的ROC曲线和P/R曲线
我们将MSDeepFilter的模型、AdaBoosting和SVM应用于训练集、交叉验证集和测试集上,得到ROC曲线。ROC图中,横轴为假阳性率,纵轴为真阳性率,曲线下面积(AUC)越大代表模型分类性能越好。在训练集、交叉验证集和测试集上,MSDeepFilter的模型的ROC曲线包裹住了其他所有模型的ROC曲线(图7),且它的测试集AUC、训练集AUC和交叉验证集AUC分别为0.9873、0.9979和0.9864,为所有模型中对应指标的最高值(表3),代表MSDeepFilter的模型在训练集、交叉验证集和测试集上的性能表现最好。
 注:不同颜色的曲线代表不同模型的性能表现。红色的线代表MSDeepFilter,绿色的线代表AdaBoosting,紫色线代表SVM。(A) 训练集上3个模型的ROC曲线图;(B) 交叉验证集上3个模型的ROC曲线图;(C) 测试集上3个模型的ROC曲线图。
注:不同颜色的曲线代表不同模型的性能表现。红色的线代表MSDeepFilter,绿色的线代表AdaBoosting,紫色线代表SVM。(A) 训练集上3个模型的ROC曲线图;(B) 交叉验证集上3个模型的ROC曲线图;(C) 测试集上3个模型的ROC曲线图。
Figure 7. ROC curves of different models
图7. 模型的ROC曲线图
4.3. 模型性能评价指标
我们将三个模型应用于训练集、交叉验证集和测试集上,获取得到的性能指标如表3所示。

Table 3. Performance indicators of models
表3. 模型的性能指标
注:AUC on Testing Set代表模型应用在测试集上得到的AUC值,同理,AUC on Training Set和AUC on Validation Set分别代表模型应用在训练和交叉验证集上得到的AUC值;黑色加粗代表每个指标里的最高值。
表3记录了训练测试中三种模型的七个性能指标。从中可以发现MSDeepFilter的七个指标中六个指标高于0.9,其中测试集AUC、训练集AUC、交叉验证集AUC三个指标高于0.95。支持向量机在测试集上的召回率(Recall)、F1分数、准确率(ACC)分别只有0.7608、0.8286、0.8367,表现较差;AdaBoosting算法在三个数据集上的AUC值分别为0.9652、0.9735、0.9641,但是精确率和准确率分别为0.9087和0.9057,低于MSDeepFilter的模型表现。
MSDeepFilter的模型的七个指标中五个指标:测试集AUC、训练集AUC、交叉验证集AUC、精确率和准确率分别为0.9873、0.9979、0.9864、0.9729、0.9317,均为对应指标中的最高值,且它的召回率和F1分数分别为0.8934和0.9315,所以综合考虑多指标,在三个模型中,MSDeepFilter的模型分类性能最优。
4.4. 模型的概率分布直方图以及对比
二分类问题的结果输出概率分布图中,两侧分布的直方越高,中间部分直方图越低,代表对两类样本的分辨能力越强。
MSDeepFilter的模型预测概率直方主要分布于两侧,对正负样本具有优秀的分辨能力;支持向量机模型预测概率直方主要分布于两侧,中间分布直方略低于MSDeepFilter,正负样本区分能力高。但是0~0.1区间分布占总数约51.7%,分布呈现负样本更多,与数据集真实标签正样本分布更多的事实不符(图8(B));AdaBoosting模型概率直方集中于0.4~0.6区间(图8(C)),虽然模型准确率达到0.9057 (表3),但从概率直方分布发现,AdaBoosting模型对两类样本的分辨能力差(图8(C))。
 注:图里所有红色直方均代表MSDeepFilter的概率分布。(A) 红色代表MSDeepFilter的分类概率分布直方;(B) 深蓝色为机器学习模型支持向量机(SVM)的分类概率分布直方;(C) 深蓝色为机器学习模型AdaBoosting的分类概率分布直方。
注:图里所有红色直方均代表MSDeepFilter的概率分布。(A) 红色代表MSDeepFilter的分类概率分布直方;(B) 深蓝色为机器学习模型支持向量机(SVM)的分类概率分布直方;(C) 深蓝色为机器学习模型AdaBoosting的分类概率分布直方。
Figure 8. Histogram of model probability distribution
图8. 模型概率分布直方图
综合上述分析,在三个模型中,MSDeepFilter的模型在区间0.1~0.9中的直方分布最少,对两类样本的分辨能力最强,性能表现最好。
5. 讨论与总结
本文基于深度学习设计了神经网络作为算法MSDeepFilter的模型,可筛选出DIA-NN输出结果中的高置信度肽段。MSDeepFilter先提取DIA实验数据中肽段对应的碎片子离子XICs进行归一化处理,然后利用深度神经网络对XICs进行分类,最后筛选出高置信度肽段。
在交叉验证数据集和测试集上,MSDeepFilter的模型性能优于传统机器学习算法AdaBoosting和支持向量机SVM。然而,由于DIA数据本身极其复杂和高通量的特点,该模型仍有很多可以完善的地方。例如,数据预处理的方法可以根据不同的质谱仪进行设计;测试数据可以加入更多质谱生产厂商的质谱仪处理的DIA数据;神经网络的结构可以设计更多不同类型的网络。
基金项目
本研究由国家自然科学基金(项目编号:12090052,11874310)提供资助。