1. 引言
4-取代-1,2,5-噁二唑甲酸作为重要的结构模块在药物化学领域有着重要应用价值。例如:咪唑[1,2-b]哒嗪IL-17A抑制剂(1)是以4-环丙基-1,2,5-噁二唑甲酸为原料合成的,其在治疗银屑病、脊柱关节炎、类风湿性关节炎和多发性硬化方面起到重要作用 [1] 。再如,以4-甲基-1,2,5-噁二唑甲酸为基础的抗结核化合物作为有效和独特的MTB抑制剂(2),可作为开发抗结核新药的有前景的先导化合物 [2] 。再如,ADAMTS7拮抗剂替代品(3)以4-甲基-1,2,5-噁二唑甲酸为原料,其单独或联合使用用于疾病的治疗或预防,特别是在心血管疾病,包括动脉粥样硬化、冠状动脉疾病、周围血管疾病等方面发挥着重要作用 [3] 。
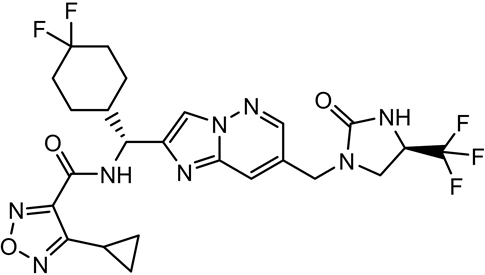
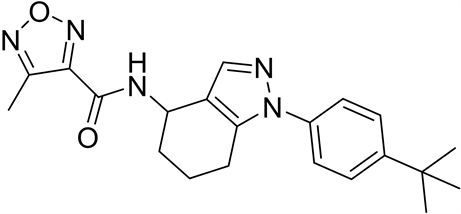
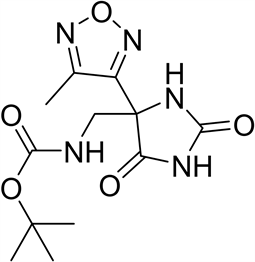
(1) (2) (3)
现有的常规合成4-环丙基-1,2,5-噁二唑-3-甲酸方法有三种:1) (1E,2E)-1-环丙基丙烷-1,2-二酮二肟与琥珀酸酐环化后,与高锰酸钾反应氧化甲基,得到目标产物 [4] ;2) 3-环丙基-3-氧代-丙酸乙酯经重氮化后环化得到目标产物 [1] ;3) (E)-3-环丙基丙烯醛先环化后琼斯氧化得到目标产物 [5] 。上述合成4-环丙基-1,2,5-噁二唑-3-甲酸的常规方法普遍存在以下问题:使用氧化剂(高锰酸钾、硝酸钠),容易产生爆炸隐患,区域选择性差,反应难以控制,总收率低,不适合大规模制备或工业化生产。
在当前倡导环保、资源可持续利用的理念下,制药行业迫切需要新的化学反应技术代替传统的间歇式化学反应技术 [6] 。与传统的间歇式化学反应技术不同,连续流化学技术是以微反应器为基础的微化工技术,具有移动化、微型化和绿色化等特征,是一种非常有前途的、可替代间歇式化学反应的新技术 [7] 。微反应器是一种连续流动的管道式反应器,其管道内径直径介于微米和毫米之间,相对于常规反应器而言,其比表面面积非常大,具有极好的换热和传质效率,可以使原料按精确的配比瞬间混合,有利于提高产品的产率,能满足绿色制造的时代要求 [7] 。故本研究提供了一种绿色的、条件温和、收率较高、降低安全隐患的4-环丙基-1,2,5-噁二唑-3-甲酸的连续流合成方法,扩大连续流化学技术的适用范围,为推动创新药物开发领域的绿色发展提供潜在的途径。
2. 实验
2.1. 原料和仪器
3-环丙基-3-氧代-丙酸乙酯(上海毕得医药科技有限公司),盐酸羟胺(萨恩化学技术有限公司),无水乙酸钠(特优级,上海泰坦科技股份有限公司),乙醇(分析纯AR,上海泰坦科技股份有限公司),亚硝酸钠(分析纯AR,国药集团化学试剂有限公司),盐酸(36%,国药集团化学试剂有限公司),Ethylene Glycol (99%,上海泰坦科技股份有限公司),氢氧化钠(分析纯AR,上海泰坦科技股份有限公司),无水硫酸钠(分析纯AR,上海泰坦科技股份有限公司)。
旋转蒸发仪(郑州科泰实验设备有限公司),核磁共振色谱仪(BrukerAvance III 400MH)。
2.2. 反应过程
2.2.1. 3-环丙基异恶唑-5(4H)酮常规合成路线
3-环丙基异恶唑-5(4H)酮合成反应式见图1。
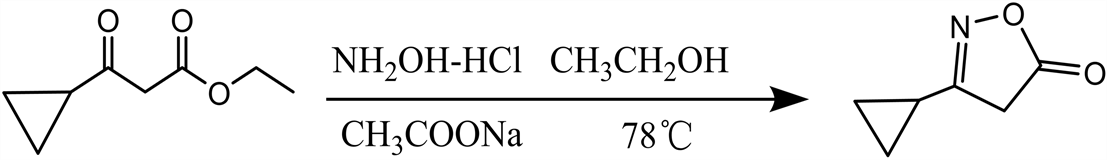
Figure 1. 3-cyclopropylisooxazol-5(4H) ketone synthesis route
图1. 3-环丙基异恶唑-5(4H)酮合成反应式
2.2.2. 3-环丙基异恶唑-5(4H)酮的制备方法
将醋酸钠(0.788 g,9.6 mmol)加入15mL乙醇中,再分批加入盐酸羟胺(0.667 g,9.6 mmol),搅拌至无气泡放出后,将3-环丙基-3-氧代-丙酸乙酯(1 g,6.4 mmol)加入到上述反应液中,控温78℃回流装置反应30 min。反应完毕后,反应液用乙酸乙酯(15 mL × 3)提取,合并有机层,干燥浓缩得到淡黄色油状3-环丙基异恶唑-5(4H)酮。
2.2.3. 4-环丙基-1,2,5-噁二唑-3-甲酸的连续化合成路线
4-环丙基-1,2,5-噁二唑-3-甲酸的连续化合成反应式与合成路线见图2、图3。
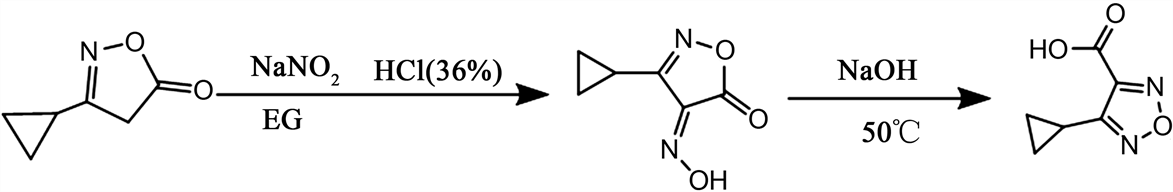
Figure 2. Reaction formula for synthesis of 4-cyclopropyl-1,2,5-oxadiazole-3-carboxylic acid
图2. 4-环丙基-1,2,5-噁二唑-3-甲酸合成反应式
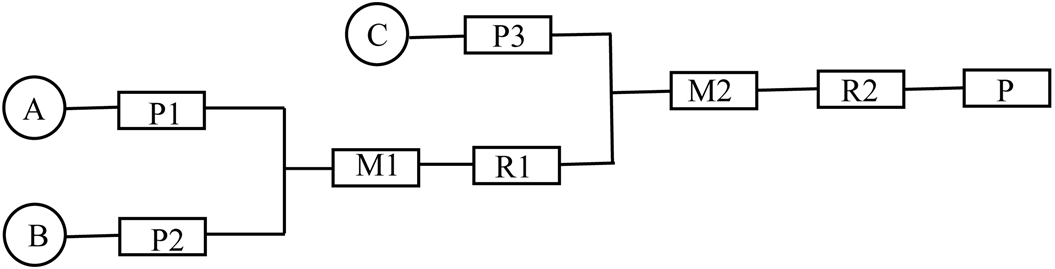
Figure 3. Line of continuous synthesis of 4-cyclopropyl-1,2,5-oxadiazole -3-carboxylic acid
图3. 4-环丙基-1,2,5-噁二唑-3-甲酸连续化合成路线
其中A为3-环丙基异恶唑-5(4H)-酮与盐酸的混合溶液、B为亚硝酸钠溶液、C为氢氧化钠溶液;M1、M2为预混模块;P1、P2、P3为计量泵;R1、R2为反应模块;P为产品收集模块。
2.2.4. 4-环丙基-1,2,5-噁二唑-3-甲酸连续化合成的制备方法
3-环丙基异恶唑-5(4H)-酮(1.33 mmol/mL)与盐酸(1.33 mmol/mL)的混合溶液(流速24μL/min)和亚硝酸钠溶液(1.92 mmol/mL,流速24 μL/min)在T型混合模块M1混合(温度20℃~30℃),在管道反应模块R1中停留30分钟,随后反应结束的反应液(流速是xx μL/min)、氢氧化钠溶液(6.40 mmol/mL,流速48 μL/min)在T型混合模块M2中混合,在管道反应模块R2中停留10分钟(温度50℃),最后混合液流入收集模块P中。将混合液用水、乙酸乙酯萃取、分液、无水硫酸钠干燥有机相后旋蒸,用二氯:甲醇 = 50:1柱层析分离得到白色固体4-环丙基-1,2,5-噁二唑-3-甲酸(91 mg,0.594 mmol/mL),总收率45%。
1HNMR (400 MHz, DMSO-d6) δ: 2.40 (tt, J = 8.4, 5.2 Hz, 1 H), 1.21~1.10 (m, 2H), 1.03~0.94 (m, 2H). MS (ESI): m/zcalcd.forC6H7N2O3[M+H]+155.0,found:155.0.
13CNMR (100 MHz, DMSO-d6) δ: 159.59, 158.34, 148.37, 9.77, 4.66.
3. 结果与讨论
3.1. R1反应模块停留时间对反应程度的影响
参照2.2.4.的制备方法合成4-环丙基-1,2,5-噁二唑甲酸,实验过程中其他反应条件相同,仅改变R1反应模块停留时间,以10min为间隔依次递加,分别测定10 min、20 min、30 min、40 min、50 min下4-环丙基-1,2,5-噁二唑-3-甲酸的总收率。30 min前,随着停留时间的增加,总收率逐渐增加;30 min后,随着停留时间的增加,总收率不再增加或增加不明显。这是因为3-环丙基异恶唑-5(4H)-酮与亚硝酸钠在盐酸的作用下随着反应时间的增加,正向反应不断进行,在反应时间为30 min时产率达到最高值并趋于稳定,故选定R1反应模块停留30 min为最适停留时间。实验结果见表1。

Table 1. Influence of residence time of R1 reaction module on reaction degree
表1. R1反应模块停留时间对反应程度的影响
3.2. 氢氧化钠溶液的通入流速对反应程度的影响
参照2.2.4.的制备方法合成4-环丙基-1,2,5-噁二唑甲酸,实验过程中其他反应条件相同,仅改变氢氧化钠溶液的通入流速,分别测定24 uL/min、48 uL/min、72 uL/min、96 uL/min下4-环丙基-1,2,5-噁二唑-3-甲酸的总收率。在固定3-环丙基异恶唑-5(4H)-酮与盐酸的混合溶液和亚硝酸钠溶液的流速为24 μL/min (混合后管道流速接近48 μL/min)的前提下,氢氧化钠溶液流速为24 uL/min时,因流速过慢,和R1混合溶液反应不充分,产物中会有大量3-环丙基异恶唑-5(4H)-酮存在,难以分离,产率低;流速为72 uL/min、96 uL/min时,因流速过快,和R1混合溶液反应时间减少,反应程度降低,产率低,且消耗大量氢氧化钠溶液,造成浪费;流速为48 uL/min时,氢氧化钠溶液与R1混合溶液可达到近似1:1的反应配比,反应程度最高,产率最高。故选定48 uL/min作为氢氧化钠溶液的通入流速。实验结果见表2。
Table 2. Influence of inlet velocity of sodium hydroxide solution on reaction degree
表2. 氢氧化钠溶液的通入流速对反应程度的影响
3.3. R2反应模块温度对反应程度的影响
参照2.2.4.的制备方法合成4-环丙基-1,2,5-噁二唑甲酸,实验过程中其他反应条件相同,仅改变R2反应模块的温度,分别测定反应温度为30℃、40℃、50℃、60℃、70℃下4-环丙基-1,2,5-噁二唑-3-甲酸的总收率。从30℃到50℃,随着温度的升高,反应不断正向进行,总收率增加;从50℃到70℃,温度太高导致副反应的发生,使总收率不再增加,反而下降,故选定50℃作为R2模块最适反应温度。实验结果见表3。
Table 3. Influence of temperature of R2 reaction module on reaction degree
表3. R2反应模块温度对反应程度的影响
4. 结论
本文以3-环丙基-3-氧代-丙酸乙酯为原料,经一步常规反应制备3-环丙基异恶唑-5(4H)酮中间体,并以3-环丙基异恶唑-5(4H)酮为原料连续化制备4-环丙基-1,2,5-噁二唑-3-甲酸,并对部分关键连续化合成条件进行探究,确定R1反应模块最适停留时间为30 min,氢氧化钠溶液的通入流速为48 μL/min,R2反应模块最适温度为50℃,收率为45%,方法安全可控在常压下反应,初步解决了现有制备方法容易产生爆炸等安全隐患,不适合大规模制备或工业化生产的技术问题。
致谢
感谢江南大学基金支持,研究和论文顺利完成。
基金项目
江苏省大学生创新创业训练计划一般项目(202210295185Y)。