1. 引言
分析化学是关于化学物质(原子和分子,以下简称“物质”)存在的科学。其方法多种多样,但都要用多个标样绘制其信息量I与其含量C的“校准曲线”,作线性回归确立线性方程 [1] - [7] 。不仅程序繁杂,而且测定结果可靠性无法保证,不确定度评定难以实施。作者发现,在每个方法实施过程,遵循“精密度法则”,即“保持各个因素对同一测定系列各个样品影响的一致性”,可以确保测定系列的“校准曲线”高度直线性,具有自己的直线方程,无需再做线性回归,确立线性方程。而且重复性“小样本”近似正态分布,最大残差容易确定,测定结果可靠性能得到保证,不确定度也可在其A类标准不确定度公式基础上,采用A类评定方法,建立起扩展不确定度的数学模型,既简便又切合实际。
2. “精密度法则”的提出
“精密度”是“在规定条件下,对同一或类似被测对象重复测量所得示值或测得值间的一致程度” [8] 。各种化学分析方法,都必然受到实施过程中“人、机、料、法、环”因素的干扰,破坏了样本各个测得值之间的一致性。为此,作者想到了“保持各个因素对同一测定系列各个样品影响的一致性”,并称其为“精密度法则” [9] 。具体细节如下。
2.1. 人的因素控制
同一测量系列的测定可以多人协作,但同一测定系列各个样品的同一程序(如称量或溶样等)必须由同一人员完成。
2.2. 机械因素控制
分析化学的机械有太平、容器、仪器等。必须保持它们在同一测定系列测试中的同一性。
2.3. 物料因素控制
首先必须保证标准物质要与被测试样的同一性。其次是保证同一测定系列所用试剂、溶剂、溶液等的同一性。
2.4. 方法因素控制
这里是指测定方法实施的方式、方法。比如标准物和样品制备的方式方法、信息量采集的方式方法等。要保持它们在同一测量系列中的同一性。
2.5. 环境因素控制
环境因素主要是方法实施过程所处环境的污染程度、温度、湿度、样品完成测定的时间等,要保持它们在同一测量系列中的同一性。
3. “精密度法则”揭示了分析化学的基本原理
分析化学各种测定方法都存在“人、机、料、法、环”影响因素的干扰,只有遵循“精密度法则”,即“保持各个因素对同一测定系列各个样品影响的一致性”,使它们对同一测定系列各个样品的影响趋于一致,测定结果才能只显出物质信息量与其含量之间的关系。
3.1. “精密度法则”条件下锰的光度法测定
选取同一碳钢系列的锰标样含量(%):0.19、0.48、0.77、0.96、1.40、1.78、2.02。
所有试样要由同一人在同一天平的同一量程(天平的不同量程的产生的称量误差是不同的),称取同样0.2000 g;再由同一人用同一量杯加硝酸(1 + 4) 30 mL;再由同一人加热溶解,并以同一方式流水冷却至室温;再由同一人在同一100 mL容量瓶中加水(必须是蒸馏水!)定容至100 mL,作为样品“母液”;再由同一人用同一10 mL刻度移液管,移取10 mL“母液”置于锥形瓶中;再由同一人用同一5 mL刻度移液管,分别加入硝酸银溶液(1%)、过硫酸铵溶液(5%)各5 mL;再由同一人在80℃~100℃水浴中摇动加热40 s,取出后,流水冷却至室温;30 min内,由同一人在721光度计上用同一只1 cm比色皿,以水作参比,在520 nm (或540 nm)处测定
吸光度。表1列举了用同一只1 cm比色皿和3个不同1 cm比色皿的测定结果。

Table 1. Absorption of manganese standard sample at 520 nm
表1. 锰标准样品520 nm的吸光度
根据表1中同一个1 cm比色皿数据绘制锰“校准曲线”的几何图形(图1)。(3个不同1 cm比色皿的“校准曲线”也是同一条,但其直线性要比同一个比色皿的差。表明遵循“精密度法则”与否,对于保证“校准曲线”的直线性至关重要!)
首先,从图1可看出,由于严格遵循“精密度法则”,所有标准样品的坐标点,几乎全部都落在同一条“校准曲线”上。这种特性不是偶然,因为无论何人何时何地,只要严格遵循“精密度法则”,都可以得到如图1的“校准曲线”。而表1数据表明,仅在最后测定中用了3个不同的1 cm比色皿,“校准曲线”虽然不变,但其中几个样品的坐标点却偏离了“校准曲线”。
其次是,“校准曲线”不通过“0”点。在无法测得一个原子或一个分子信息量之前,其“校准曲线”只能不同程度地逼近“0”点,而不会到达“0”点,所以各种方法“校准曲线”的数学方程都不会是“点斜式”,而是“两点式”:
(1)
从图1还可看出,
就是“校准曲线”的斜率k。如果以ΔA表示吸光度的改变量,ΔC表示相应物质含量的改变量,则图1“校准曲线”还有如下方程式:
(2)
上述事实表明:遵循“精密度法则”,回避了各种影响因素对“校准曲线”的干扰,其线性方程式2才体现出了光度法的真实规律:“吸光度改变量与吸光物质含量改变量成正比”。这也正是光度法实际偏离比尔定律的根本原因。这一规律的存在表明,在遵循“精密度法则”条件下,所有样品的坐标点,都会落在CI与CII两个坐标点的连线上,所以,这条连线可替代“校准曲线”,作为样品含量Cx的“测定线”。同时根据式(1)样品含量Cx还有如下的数学模型:
(3)
同批样品测定
是同一的,以大写的K表示,则式(3)可简化为:
(4)
样品含量Cx既可从“测定线”上查得,也可由式(4)计算求得。它们都只用上、下限两个标准样品,所以统一简称作“两标样测定法”。“测定线”简化了“校准曲线”的绘制,而式(4)则是对“校准曲线”的彻底摆脱,是光度法科学性的体现,也是对光度法的重新认知。
3.2. “两标样测定法”的普适性
实际上,在无法获得一个原子或一个分子信息量之前,分析化学各种方法的“校准曲线”都只能不同程度地逼近坐标点“0”,而不能真正达到“0”。所以分析化学的“校准曲线”方程都不会是“点斜式”而只会是“两点式”。遵循“精密度法则”,同样会保障它们高度的直线性。所以光度法“校准曲线”解析所得的“两标样测定法”,对它们都是适用的,只要将其信息量吸光度A,代之于共同信息量I,将式(2)、式(4)变为如下形式:
(5)
(6)
它们表明,严格遵循“精密度法则”,各种“校准曲线”都会具有高度的直线性,它们显示了分析化学的基本原理:“物质信息量的改变量与其物质含量的改变量成正比”。
例1. 在光度法中的应用
表1中锰含量上限CII = 2.02%、AII = 0.860,下限CI = 0.19%、AI = 0.114,
。按照“两标样测定法”确定表1中吸光度分别为0.235、0.350、0.428、0.608、0.760的锰含量。结果见表2。
Table 2. Results of “Two standard sample determination method” for Manganese (%)
表2. 锰“两标样测定法”结果(%)
例2. 在“增量法” [10] 中的应用
应用本文
光度法,对含锰量低于0.19%的铁粉(标称值0.030%)的测定。
选取不含锰的纯铁粉作为CI = 0.00%,含锰量0.96%的碳钢样品作为CII,并以CII作为测定系列的“增量”。
按照本文3中的方法,称取各个试样0.2000 g,制成各自的“母液”100 ml。每种“母液”都用同一10 ml刻度移液管移取10 ml,分别置于200 ml的锥形瓶中。再用同一10 ml刻度移液管向3个锥形瓶中分别加入CII“母液”10 ml作为测定系列的“增量”。再用同一5 ml刻度移液管向3个锥形瓶中分别加入硝酸银溶液、过硫酸铵溶液各5 ml。按照本文2.3方法显色、测定各自
的吸光度。
由于所有样品都加入了10 ml CII作为测定系列的系统“增量”,测定液体积从原方法的20 ml稀释至30 ml,所以标样锰的实际含量CII = 0.96(2/3) = 0.64%。测定系列为0.00%、Cx和0.64%。3个样品的吸光度A分别为0.300、0.315、0.560,
。以其“测定线”确定Ix = 0.315的Cx = 0.037%。以式(6)计算Ix = 0.315的Cx = 0.037%。
“增量法”表明,所谓“检出限”的提法是不科学的,低于“检测限”含量,是可通过“增量法”,使它的信息量进入“信息量下限”而被测定。
例3. 在摄谱法中的应用
摄谱法的信息量I是被测元素分析线对的“黑度差”。下面是摄谱仪对含Cr18.41%、含Ni8.09%的不锈钢样品的测定结果 [11] 。
Cr的分析线对为Cr2590/Fe2588。测定结果列于表3,“校准曲线”省略。
Table 3. Cr standard sample series “blackness difference”
表3. Cr标样系列“黑度差”
CII = 19.96%、III = −0.524,CI = 13.20%、II = −0.850,
。样品“黑度差”Ix = −0.586相应Cr含量从“测定线”查得18.68%,由式(6)测得18.68%;
Ni的分析线对Ni3510/Fe3498。测定结果列于表4,“校准曲线”省略。
Table 4. Ni standard sample series “blackness difference”
表4. Ni标样系列“黑度差”
标样CII = 12.51%、III = 0.534,CI = 7.11%、II = 0.110,
。样品“黑度差”Ix = 0.197相应Ni含量Cx从“测定线”查得8.33%,由式(6)测得8.22%。
例4. 在滴定法中的应用
因为黄铜样品的含铜量范围很宽(56%~96%),采用滴定度换算误差过大,甚至可达1.2%,而采用“校准曲线”法,则可把误差降至0.1%以下 [12] 。
6个含铜标样铜含量(%):57.19、61.96、74.45、88.34、95.66、99.99。
称取0.2000 g试样加盐酸5 ml、过氧化氢3 ml,低温溶解,流水冷却至室温,在200 ml容量瓶中加水定容。用10 ml移液管移取10 ml试样于锥形瓶中(每个试样移取两个平行样品),加氟化铵1.5 g,摇匀。加碘化钾4 g,立即用0.1%硫代硫酸钠溶液滴定。呈淡黄色后加淀粉溶液(0.5%) 3 ml,继续滴定至蓝色消失色。滴定结果列于表5。
Table 5. Consumption of titrant for brass samples (ml)
表5. 黄铜样品滴定剂耗量(ml)
“校准曲线”(省略)。这里只展现它的“两标样测定法”应用:上限CII = 99.99%、III = 46.18 (ml),下限CI = 57.19%、II = 27.14 (ml),
。
样品滴定液用量Ix = 34.73 (ml)的相应铜含量Cx,从“测定线”查得74.3%,由式(6)计算得74.27%;样品滴定液用量Ix = 44.34 (ml)的相应铜含量Cx,从“测定线”查得95.8%,由式(6)计算得95.89%。
以上实例表明分析化学不再是一项依赖于“校准曲线”的实验性技术,而是一门具有自己的基本原理、跨物理和化学两大学科的基础科学。
3.3. “两标样测定法”的实际应用
“测定线”,简化了“校准曲线”的绘制,而式(6)则是对“校准曲线”的彻底摆脱。从图1可以看出Cx处于
中的CII、CI之间。CII、CI并非必须是“校准曲线”的上、下限,只要是可包含Cx的上、下限。比如,用光度法测定含锰量在0.5%~1%的试样。即可选锰含量低于0.5%的标样作为所用区间的下限,选含量 > 1%的标样作为所用的上限。例如,选CI = 0.48%、II = 0.235,CII = 1.40%、III = 0.608,相应的K = 2.47。按照式(6) Ix = 0.350的相应锰含量Cx = 0.76%。实际表2中吸光度A = 0.350的样品含量为0.77%,基本与以下限CI = 0.19%,上限CII = 2.02%的结果是相同的。
对于含量Cx低于下限的试样,则可采用 “增量法”。只要选一个CI = 0.00%和一个含量大于Cx的标样作为其上限,并以其作为测定系列的“增量”即可。
式(6)的实用性,表明方法研究者,重要的是,提供方法可应用信息量的上、下限,而不是可测定物质含量的上、下限。因为同一信息量的上、下限,既可用于测定微含量,也可用于测定高含量。
4. “精密度法则”确保了样本在极差R区间内的正态分布
概率论的中心极限定理表明,受一系列独立因素影响的随机变量,如果各个变量Xi的变化对其总合的影响“都很微小”,则变量Xi的分布极限是正态分布 [13] [14] 。遵循“精密度法则”,被测量μ的同一样本的各个测得值Xi就会“如出一模”,它们各自的变化对其总合的影响必然“都很微小”。所以,测得值Xi的分布极限就是正态分布。即μ的“大样本”均值
会与μ重合于样本极差R的中心,各个测得值Xi会密度逐渐降低的、均称的分布于μ(
)两侧的0.5R内(图2)。样本测得值在极值R区间内的正态分布,为系统误差和统计离群值的发现提供了有利条件。因为它们的存在,都会破坏这种分布的均称性 [15] 。系统误差可通过标准样品的测定而确定,统计离群值则是大于最大值(
)或小于最小值(
)值,即超出(
)的测得值。
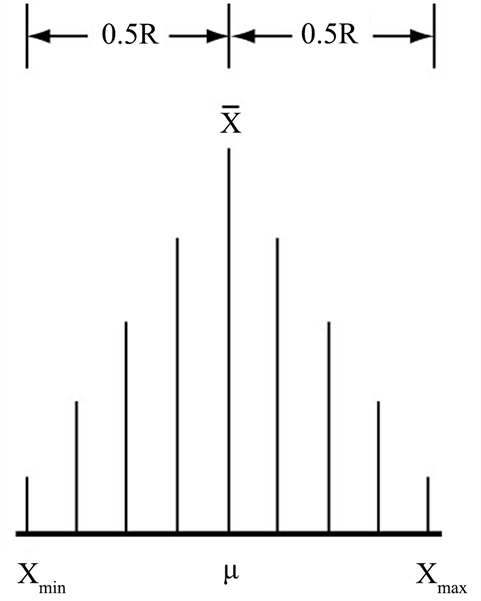
Figure 2. Normal distribution of large samples
图2. 大样本正态分布
“大样本”是正态分布,“小样本”必然近似正态分布。它的各个测得值Xi也会较均称的分布于其均值
的两侧,系统误差和统计离群值同样会破坏这种均称性。系统误差同样可通过对标准样品的测定而确定。由于“小样本”大于μ的均值
处在μ右侧距最小值的eR处,小于μ的均值
处在μ左侧距最大值的eR处,统计离群值就是超出(
)区间的测得值。从图2可知,eR值处于0.5R~R之间,根据“小样本”近似正态分布,参照正态样本统计离群值判断中的格拉布斯法 [16] ,eR值就容易确定。作者在大量n > 5“小样本”中应用“折半法”,选择超出(
)区间的eR值。最终确定其为0.65R,即超出(
)区间的测得值,就是应剔除的统计离群值。
排除了系统误差和统计离群值,就提高了测定结果的可靠性。比如,钢中锰(标称值0.66%)的光度法测定结果(%):0.66、0.67、0.68、0.66、0.68、0.67、0.68、0.67、0.74,均值
,极差R = 0.08%。9个测得值6个小于均值,3个大于等于均值,分布明显不均称,表明测得值中含有系统误差或统计离群值,所以均值
不可靠。系统误差排除程序繁琐,可先判断统计离群值的存在。(
)区间为0.63%~0.73%,测得值0.74%超出了该区间,属于统计离群值,应予剔除。剔除0.74%后,均值
,极差R = 0.02%。8个测得值2个小于均值,3个大于均值,基本均称,表明不再有系统误差。其(
)为0.66%~0.68%,8个测得值均在其间。表明也不存在统计离群值,所以均值
是可靠的。实际上它比0.68%更贴近标称值0.66%。
5. “精密度法则”创造了分析化学样本测量不确定度评定的最佳条件
参与分析化学方法实施过程的“人、机、料、法、环”,都会影响它测量结果的不确定度。这些因素各自引入测量结果的不确定度分量很难估计,所以“合成法”评定结果常常不切合实际。惟有遵循“精密度法则”,保证样本测得值的重复性,才可以重复性测量均值
的A类标准不确定度公式为基础,采用A类评定方法评定。
规范JJF1059.1-2012 [17] (以下简称“规范”) 4.3.2.6指出,被测量
重复性测量均值
的A类标准不确定度公式为:
(7)
统计学指出,
是“小样本”均值
的实验标准差。它表明
落在
和
两个区间的概率分别为0.9545%和0.9973% [18] 。“规范”之4.5指出,这两个包含区间的半宽度分别是
的两个扩展不确定度:
(8)
(9)
S是样本观察值Xi的标准差。根据“规范”4.3.2.3,近似正态分布条件下,S可按照极差法
评估。“规范”4.3.2.6要求,“小样本”容量n应取10,所以
,
。将
代入式(8)、式(9),得到均值
的扩展不确定度的数学模型:
(10)
(11)
它们表明,“小样本”均值
的不确定度与样本极差R成正比,与样本容量
成反比。而且因为
,所以它们都应小于0.5R,这是判断不确定度合格与否的简单尺度。实践证明,虽然它们是在样本容量为10下确定的,但其适用于n > 5的重复性测量。而且它是“规范”中“在任何给定被测量的测量中实际可达到的最小测量不确定度”。本法不仅比“合成法”简便,而且应用效果切合实际 [19] 。
例1. 样品茶叶中敌敌畏残留量10次独立测定结果(mg/kg):0.055、0.053、0.050、0.049、0.045、0.054、0.046、0.052、0.049、0.042,均值
,极差R = 0.013。10个测得值5个大于等于均值,5个小于均值,基本均衡,所以不存(
)在系统误差。(
)区间0.042~0.058,10个测得值均在其间,不存在统计离群值,均值
有效。其
,检测结果为(0.050 ± 0.003) mg/kg、k = 2。而合成法
[20] ,几乎是
的3倍,而且大于0.5R表明它是不符合不确定度要求的。
例2. X-3600X荧光光谱仪检测含金量(99.994%)的千足金样品10次重复测得值(%):99.99、99.99、99.99、99.99、99.95、99.96、99.99、99.96、99.95、99.99、99.98,均值
,极差R = 0.04%。10个测得值分布基本均衡,也都没有超出(
)区间,表明没有系统误差和统计离群值;根据式(10)
,测定结果为(99.98 ± 0.01)%、kp = 2,结果与样品实际含金量相符。而合成法的
(是极差的13倍!)、kp = 2 [21] ,测定结果(99.98 ± 0.52)%、kp = 2,该结果已经远远超出了应当剔除的统计离群值(
),实际上它是对实际的检测结果的否定,也是对千足金样品的否定。
例3. 差示扫描量热法对标准物纯度为(99.95 ± 0.03)%苯的8个重复测得值(%):99.93、99.93、99.94、99.97、99.98、9998、99.98、99.96,均值为99.96%,极差R = 0.05%。8个测得值分布均衡,而且都没有超出它的(
)区间,表明没有系统误差和统计离群值。根据式(11)
,测定结果为(99.96 ± 0.02)%、kp = 3,符合样品实际。而依据JJF1059-1999的合成标准不确定度计算,置信概率为0.99时,扩展不确定度U = 0.11%、kp = 1.71 [22] 。测定结果为(99.96 ± 0.11)%、kp = 1.71。该结果同样超出了应当剔除的统计离群值,同样是对实际测定结果的否定,也是对差示扫描量热法的否定。
6. 结论与建议
1) “精密度法则”不仅展现了“物质吸光规律”,揭开了光度法实际与比尔定律不符的根本原因,而且也呈显了分析化学的基本原理,导出了“两标样测定法”,使其摆脱了对“校准曲线”的依赖。不仅简化了操作,还展现了它既可测定微含量,也可测定高含量的科学性;
2) 确保了分析化学样本测定结果在R区间内的正态分布,为排除系统误差和统计离群值创造了条件,保证了测定结果的可靠性;
3) 确保了样本测得值的重复性,为应用其A类不确定度公式,采用其A类评定方法创造了条件,导出了“小样本”测量结果的U95和U99数学模型。达到了“在任何给定被测量的测量中实际可达到的最小测量不确定度”。不仅比合成法简便,而且应用效果切合实际 [19] ;
4) 分析化学各种方法中,必然存在着“人、机、料、法、环”各种因素的干扰。只有遵循“精密度法则”,才可凸现出它们的基本原理,并具备了保证测定结果可靠性的条件和测定结果的不确定度数学模型,成为了完善的“关于化学物质存在的基础科学”。所以,作者认为它是分析化学必备的基础技术。