1. 引言
系统性红斑狼疮(SLE)是一种复杂的、多系统的、慢性复发性免疫疾病,具有多种临床表现和显著的发病率和死亡率 [1] 。2022年SLE发病率和流行率的报告显示,全球SLE发病率为0.0051%,新确诊人群每年大约在40万人;全球患病率是4.37%,而受影响人群每年在341万人 [2] 。因此,有必要寻找有效的生物标志物进行早期检测和预防治疗。
目前,许多研究表明表观遗传修饰在包括SLE在内的一些自身免疫性/炎症疾病的发病机制中起重要作用 [3] 。DNA甲基化最主要的功能是调节附近的基因表达 [4] [5] 。SLE中异常的DNA甲基化导致免疫失调 [6] 。本研究基于GEO数据库,使用统计软件R对基因表达数据、DNA甲基化数据进行差异分析,寻找被DNA甲基化影响的基因,进一步分析异常甲基化差异表达基因的生物功能,并通过蛋白质–蛋白质相互作用(PPI)网络分析,此外,我们用其他GEO数据集对hub基因的表达和甲基化水平进行验证。
2. 材料与方法
2.1. 数据来源
基因表达综合数据库(GEO, https://www.ncbi.nlm.nih.gov/geo/)是一个在线数据库,提供全面的基因谱和测序数据。在GEO数据库中,我们检索了外周血单核细胞的基因表达谱数据集GSE81622和DNA甲基化谱数据集GSE82218,其中包含30名中国系统性红斑狼疮患者(其中有15名患有狼疮肾炎,15名不患有狼疮肾炎)和25名健康对照组,DNA甲基化谱数据集GSE82218基于GPL13534平台(Illumina HumanMethylation450 BeadChip);基因表达谱数据集GSE81622基于GPL10588平台(Illumina HumanHT-12 V4.0 expression beadchip)。
2.2. 差异甲基化和表达基因
对于多组学数据进行差异分析鉴定SLE相关基因。使用R软件中的limma包分析SLE的基因表达数据,limma是一种基于广义线性模型的差异表达筛选方法。设置
,
为差异有显著意义。ChAMP包适用于450 K和850 K的甲基化数据分析,则使用ChAMP包识别差异甲基化基因,
,
被认为是“差异标准”。
2.3. 功能富集分析
GO通路富集分析包括细胞成分(cellular components, CC)、生物过程(biological process, BP)、分子功能(molecular function, MF)。京都基因与基因组百科全书(Kyoto Encyclopedia of Genes and Genomes, KEGG)是一个数据库资源,用于从通过高通量实验技术生成的大规模分子数据集中了解高级功能和生物系统。DAVID是一种在线功能注释工具,用于大型基因的富集分析 [7] ,本文使用DAVID在线分析,富集分析的显著性用
的标准。
2.4. PPI网络的构建
蛋白质–蛋白质相互作用网络(Protein-Protein Interaction Networks, PPI)是由蛋白通过彼此之间相互作用构成,可以用来识别关键基因和重要模块 [8] 。检索相互作用基因的搜索工具(STRING, https://string-db.org)数据库是一个全球资源,提供了PPI的验证和预测信息。本文基于STRING数据库构建了受DNA甲基化影响的差异基因(mDEG)的PPI网络,使用Cytoscape中的CytoHubba插件确定关键基因。
2.5. 相关性分析
疾病的发生和发展是极其复杂的,它是由多种调节因子共同作用的,所以,有必要探究DNA甲基化与基因表达之间的关联程度。相关系数能够反映变量之间相关关系程度的统计指标,它的取值范围是[−1,1],当取值为0时表示不相关,当取值为[−1,0)时表示负相关,取值为[0,1]时,则表示正相关。
2.6. 使用其他GEO数据集验证hub基因
我们用其他数据集验证了枢纽基因的表达和甲基化水平。基因表达数据集GSE45291包含有292个SLE和20个对照样品,用于验证枢纽基因的表达水平。用DNA甲基化数据集GSE96879验证枢纽基因的基因甲基化水平,包括57个SLE和33个对照组。当比较两组时,使用Student t检验进行统计学显著性,并且
被认为是统计学显著的。并利用pROC包绘制ROC曲线,曲线下面积AUC值越大表示诊断效果越好。
2.7. 关键基因-miRNA网络的构建
我们使用NetworkAnalyst (PMID: 30931480)数据库来预测关键基因的miRNA。将hub基因及其miRNA整合到调控网络中,并使用Cytoscape软件进行可视化。使用mirPath v3.0工具 (https://dianalab.e-ce.uth.gr/html/mirpathv3/index.php?r=mirpath)的基因本体(GO)分析,我们研究了miRNA 的作用,选择p值< 0.05的显著项。
3. 结果
3.1. DNA甲基化分析
在甲基化数据集GSE82218中,在SLE和对照组之间筛选出820个差异甲基化位点(DMP),包括496个高甲基化位点和324个低甲基化位点,火山图如图1(a)。CpG位点分布在TSS1500、TSS200、5'UTR、1stExon、body和3'UTR等不同基因区域。如图1(b)所示,在1stExon区域有29个CpG位点的的差异甲基化基因(DMGs),在3'UTR区域有36个CpG位点的DMGs,在5'UTR区域有91个CpG位点的DMGs,在TSS1500区域有102个CpG位点的DMGs,在TSS200区域有53个CpG位点的DMGs,在Body区域有363个CpG位点的DMGs。高甲基化基因和低甲基化基因不同区域的CpG位点分布相似,近50%的甲基化改变发生在Body区域。
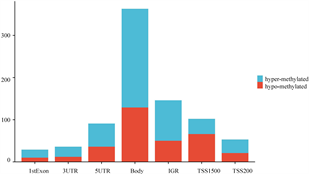
 (a) DMG火山图 (b) DMG的功能位置
(a) DMG火山图 (b) DMG的功能位置
Figure 1. DNA methylation analysis of SLE and control samples
图1. SLE与对照组DNA甲基化分析
3.2. 基因表达分析
在基因表达数据集GSE81622中,使用Limma包根据阈值
和
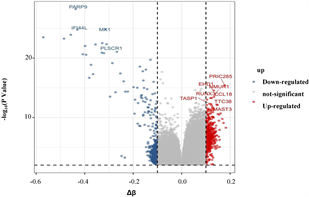
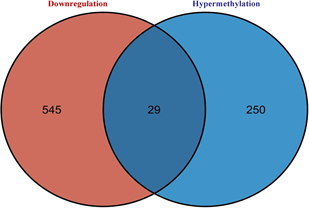
筛选差异表达基因,共鉴定出了916个差异基因(DEG),其中342个上调,574个下调(图2(a)和图2(b)) [9] 。将DEG和DMG取交集,我们发现29个基因是高甲基化低表达(Hyper-down) (图2(c)),19个基因是低甲基化高表达(Hypo-up) (图2(d))。Hyper-down基因的CpG位点按基因区域分布为:1stExon (4个基因)、5'UTR (2个基因)、Body (21个基因)、TSS1500 (4个基因)、TSS200(9个基因) (图3(a))。CpG位点分布在Hypo-up基因的不同区域,包括1stExon (3个基因)、3'UTR (1个基因)、5'UTR (5个基因)、body(8个基因)、TSS1500 (7个基因)和TSS200 (个基因) (图3(b))。结果表明,异常甲基化表达基因的CpG位点主要分布在Body区域,特别是在Hyper-down基因中。Hyper-down的CpG位点在3'UTR区域没有且在5'UTR区域数量最少,Hypo-up基因的CpG位点在3'UTR区域数量最少。
3.3. 功能富集分析
将29个hyper-down和19个hypo-up基因导入DAVID数据库进行分析,hyper-down基因在GO-生物过程(图4(a)),主要富集到免疫相关的通路和T细胞相关的通路,例如免疫反应调节细胞表面受体信号通路、免疫反应–激活细胞表面受体信号通路、T细胞活化、T细胞受体信号通路等;KEGG通路富集结果显示(图4(b)),大多数高甲基低表达基因参与自然杀伤细胞介导的细胞毒性,少数高甲基低表达基因与I型糖尿病、移植物抗宿主病和自身免疫性甲状腺疾病有关。
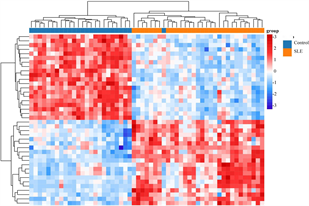
 (a) DEG的火山图 (b) DEG的热图
(a) DEG的火山图 (b) DEG的热图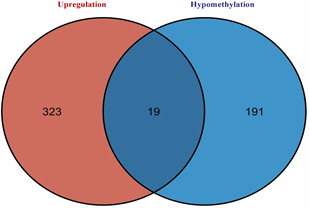
 (c) 高甲基化和低表达基因的交集 (d) 低甲基化基因和高表达基因的交集
(c) 高甲基化和低表达基因的交集 (d) 低甲基化基因和高表达基因的交集
Figure 2. Gene expression analysis of SLE and control samples
图2. SLE和对照样品的基因表达分析
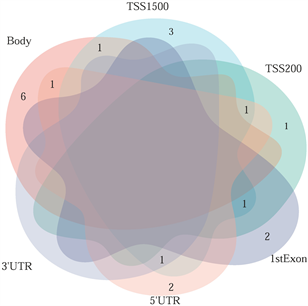
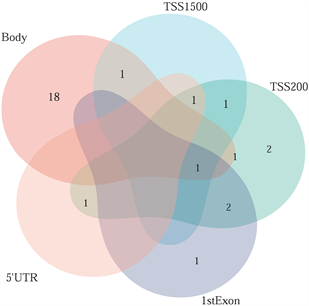 (a) 在SLE中不同区域的高甲基化低表达基因 (b) 在SLE中不同区域的低甲基化高表达基因
(a) 在SLE中不同区域的高甲基化低表达基因 (b) 在SLE中不同区域的低甲基化高表达基因
Figure 3. Abnormal methylation of differentially expressed genes based on CpG in different regions
图3. 基于不同区域CpG的异常甲基化差异表达基因
此外,hypo-up基因的GO-生物过程(图4(c)),主要与对病毒的反应、对共生体的防御反应、I型干扰素的反应等有关;KEGG分析表明(图4(d)),hypo-up主要涉及丙型肝炎、甲型流感、癌症中的转录失调、麻疹和冠状病毒病(COVID-19)。

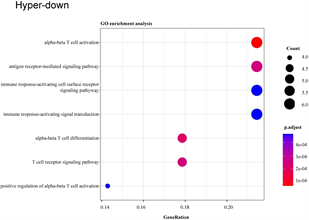 (a) and (b):hyper-down基因的GO富集分析和KEGG通路分析
(a) and (b):hyper-down基因的GO富集分析和KEGG通路分析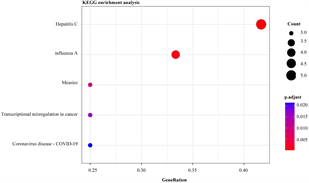
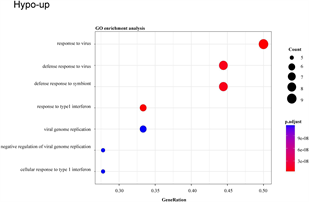 (c) and (d):hypo-up基因的GO富集分析和KEGG通路分析
(c) and (d):hypo-up基因的GO富集分析和KEGG通路分析
Figure 4. Functional enrichment analysis of the mDEGs
图4. mDEGs的功能富集分析
3.4. PPI网络分析和关键基因鉴定
使用STRING数据库分析受甲基化影响的差异基因(mDEG)的PPI网络,并用Cytoscape软件可视化,并识别关键基因。总共29个节点和29个边涉及在hyper-down的PPI网络(图5(a)),同时将连接度最高的前5个hyper-down确定为hub基因,从黄色变为红色的颜色表示蛋白质的等级,并且更深红色表示更高等级的蛋白质。结果显示,PRF1是具有最大连接度的显著基因,其次分别是ITGAL、ITK、CD247、FASLG、ZAP70、CD7、RUNX3、CD160、BCL11B (图5(b))。hypo-up基因的PPI网络包含19个节点和70个边(图5(c)),其中USP18、IFIT1、OAS3、RSAD2、OAS2都被鉴定为具有最大连接度的显著基因,其次是CMPK2、MX1、HERC5、IFI44L、IFI44 (图5(d)),最后我们将最大连接度的6个基因作为关键基因。
3.5. 甲基化特征与基因表达特征的相关性分析
Spearman相关系数的方法对甲基化数据和基因表达数据进行计算,根据相关系数大小筛选出显著相关的甲基化特征与基因表达特征,确定甲基化特征与基因表达特征的差异情况(图6)。
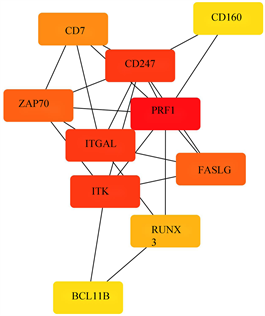
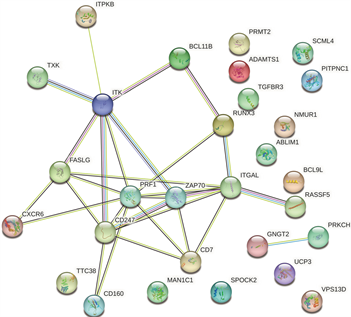 (a) hyper-down的蛋白互作网络 (b) hyper-down的hub基因
(a) hyper-down的蛋白互作网络 (b) hyper-down的hub基因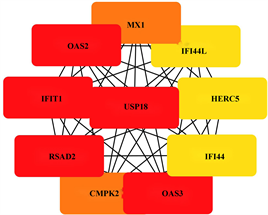
 (c) hypo-up的蛋白互作网络 (d) hypo-up的hub基因
(c) hypo-up的蛋白互作网络 (d) hypo-up的hub基因
Figure 5. PPI network analysis and hub gene selection of hyper-down genes and hypo-up genes
图5. PPI网络分析和hyper-down基因和hypo-up基因选择hub基因
从表1中可以看出,一个基因的表达异常并不仅仅和它自身的甲基化相关,也可能是其他多个基因的甲基化共同作用的效果,例如,基因USP18的表达上调可能与PRF1高甲基化以及IFIT1、OAS2、OAS3、RSAD2、USP18的低甲基化有关,这些基因的改变的很可能对SLE的发生有影响。
3.6. hub基因的验证
为了确认hub基因的甲基化和表达水平,我们分析了来自GEO数据库中的另外两个独立的数据集(GSE45291和GSE96879)。在GSE96879数据集中,SLE组PRF1甲基化水平显著高于对照组。SLE
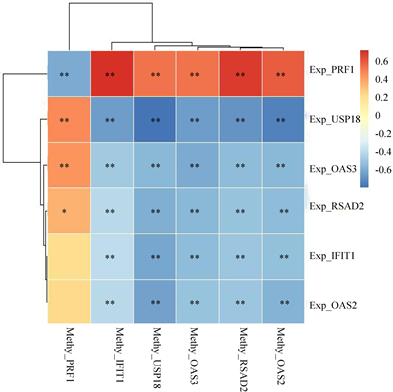
Figure 6. The speaman correlation between methylation characteristics and gene expression characteristics
图6. 甲基化特征与基因表达特征的相关关系

Table 1. Highly the spearman correlation methylation characteristics and gene expression characteristics
表1. 相关性高的甲基化特征与基因表达特征
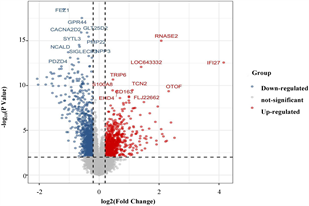
患者IFIT1、OAS2、OAS3、RSAD2、USP18基因甲基化水平显著低于正常对照组(图7(a))。在GSE 45291数据集中的mRNA水平,与对照组相比,SLE组中的PRF1基因显著下调,IFIT1、OAS2、OAS3、RSAD2、USP18均显著上调(图7(b))。这些结果进一步证明了PRF1是高甲基化状态和下调表达,以及IFIT1、OAS2、OAS3、RSAD2、USP18均是低甲基化状态上调表达。并利用pROC包绘制ROC曲线,确定hub基因的诊断价值。根据ROC曲线显示,6个基因的AUC值均在0.6以上(图8(a)和图8(b))。说明这6个基因可以成为SLE的关键基因,并有良好的诊断价值,可以确定它们在SLE中具有良好的诊断性能。
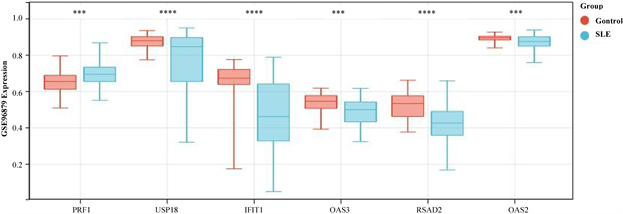 (a)在GSE96879数据集中hub基因的甲基化水平
(a)在GSE96879数据集中hub基因的甲基化水平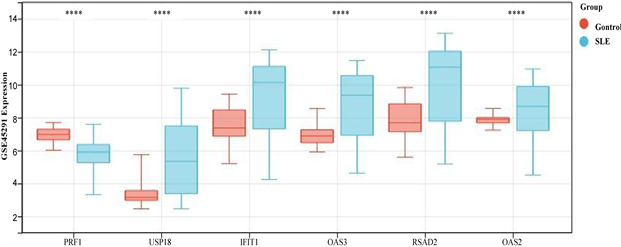 (b)在GSE45291数据集中hub基因的表达水平
(b)在GSE45291数据集中hub基因的表达水平
Figure 7. Validation expression levels and methylation levels hub genes
图7. 验证hub基因的表达水平和甲基化水平
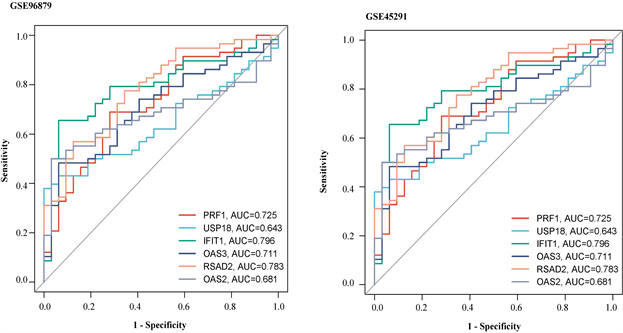 (a)在GSE96879数据集中hub基因ROC曲线 (b)在GSE45291数据集中hub基因的ROC曲线
(a)在GSE96879数据集中hub基因ROC曲线 (b)在GSE45291数据集中hub基因的ROC曲线
Figure 8. ROC curve verification of hub gene
图8. hub基因的ROC曲线验证
3.7. 共享miRNA及其作用
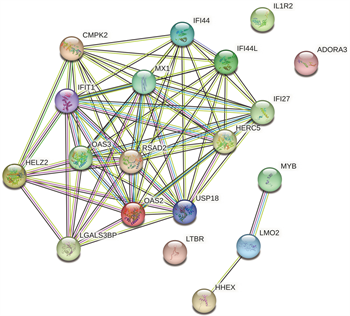
利用NetworkAnalyst数据库,通过Cytoscape软件构建了miRNA-hub基因网络,该网络包括6个中心基因、37个miRNA (图9(a)),为了便于选择重要的miRNA,网络中选择了至少5个hub基因的miRNA,有8个与关键基因共享的miRNA,分别为hsa-mir-26a-5p、hsa-mir-27a-5p、hsa-mir-129-2-3p、hsa-mir-210-3p、hsa-mir-212-3p、hsa-mir-221-3p、hsa-mir-452-5p和hsa-mir-449b-5p。最后使用mirPath对8个miRNA进行GO富集,表明这些miRNA的功能参与免疫过程、toll样受体通路(图9(b))。

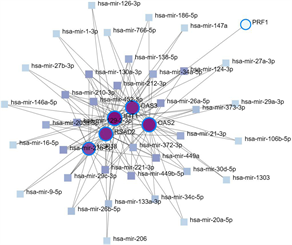 (a) 与hub基因相关的共享miRNA (b) 共享miRNA中的GO富集
(a) 与hub基因相关的共享miRNA (b) 共享miRNA中的GO富集
Figure 9. The hub genes-miRNAs network
图9. hub基因-miRNAs网络
4. 讨论
系统性红斑狼疮是一种慢性自身免疫性疾病,临床表现复杂,导致多个系统性缺陷。据统计,10万人就会有150人会发生红斑狼疮和15%患有系统性红斑狼疮患者5年内有肾功能衰竭或死亡 [10] 。SLE的发生发展受很多因素的共同影响,如基因、环境,表观遗传学和激素,而表观遗传标记的可修饰性质将成为未来治疗的关键目标,DNA甲基化是表观遗传学的一种研究充分且稳定的形式,这也引起了越来越多的关注DNA甲基化在SLE的病理生物学的重要性 [11] [12] [13] 。
在这项研究中,使用表达阵列数据集GSE81622鉴别SLE的差异基因,再分析DNA甲基化谱数据集GSE82218中的差异甲基化基因,联合基因表达和DNA甲基化数据,揭示DNA甲基化相关的48个基因(mDEG),并在我们的研究中,SLE中的大多数CpG位点被发现是低甲基化。进一步GO富集分析显示,高甲基低表达基因主要富集到免疫应答调节信号通路、免疫反应激活信号传导通路;低甲基高表达基因主要涉及病毒防御反应、I型干扰素的反应 [14] 。信号通路结果显示,高甲基低表达基因自然杀伤细胞介导的细胞毒性、白细胞跨内皮细胞迁移和T细胞受体信号通路;低甲基化高表达基因主要富集甲型流感、丙型肝炎等条目。再通过PPI互作网络分析确定上述mDEG编码蛋白的相互作用关系,并得到了6个hub基因:PRF1、IFIT1、OAS2、OAS3、RSAD2、USP18,在外周血中,它们有可能成为促进系统性红斑狼疮早期诊断的诊断标记物,并且我们DNA甲基化也参与了系统性红斑狼疮的发生。最后,共享的 miRNA,hsa-mir-26a-5p、hsa-mir-27a-5p、hsa-mir-129-2-3p、hsa-mir-210-3p、hsa-mir-212-3p、hsa-mir-221-3p、hsa-mir-452-5p和hsa-mir-449b-5p,也可以调节关键基因。
OAS2、OAS3、IFIT1均为干扰素诱导蛋白,OAS3可以降解病毒RNA,有研究表明 [15] ,长链非编码RNA-MALAT1可通过上调OAS2、OAS3促进SLE病情。IFIT家族成员通过多种机制调节免疫反应,限制病毒感染 [16] ,有文献表明,在狼疮肾炎中IFIT1的表达与足细胞损伤有关 [17] 。RSAD2是Ⅰ型IFN介导的与内质网的病毒抑制基因,且Jang [18] 等人证实RSAD2与树突状细胞的成熟有关,当RSAD2高表达可能与树突状细胞的免疫失衡有关系,又有研究早期证明树突状细胞参与的免疫调控与SLE的发病密切相关 [19] ,并且本文研究出RSAD2的高表达可能受到DNA甲基化的调控。USP18是一种干扰素信号通路抑制分子,有人研究显示 [20] USP18缺失可以影响小鼠体内调节T细胞发分化,Li等人研究表明 [21] ,SLE体内Treg水平显著下调,所以USP18通过低甲基调节表达上调可能会对SLE起重要作用。(PRF1)是溶细胞颗粒的主要溶细胞蛋白之一。淋巴细胞介导的细胞溶解的主要途径之一是将T细胞或NK细胞型的溶细胞效应淋巴细胞中所含的溶细胞颗粒分泌到靶膜上,该基因与噬血细胞性淋巴组织细胞增多症(HLH)有关,但在系统红斑狼疮中没有报道。
总之,本研究提供了系统性红斑狼疮患者DNA甲基化和基因表达之间相互作用的综合观点。结果表明,所鉴定的mDEG与免疫及相关的生物学功能、甲型流感和丙型肝炎有关。PRF1、IFIT1、OAS2、OAS3、RSAD2和USP18是SLE病理过程中涉及的核心基因,同时共享miRNA也是关键基因的调节治疗靶点。这些结果为我们更好地了解SLE的发病机制提供了基础方向上的新启示,并为SLE的诊断和治疗提供有价值的新生物标志物。
基金项目
本研究由国家自然科学基金资助项目(62063024, 61461038),中央引导地方科技发展计划(RZ2300000684)和内蒙古自治区高等学校科学研究项目(NJZY20005)提供资助。