1. 引言
乳腺癌是目前女性常见三大癌症之一,据统计2018国内乳腺癌确诊人数占女性癌症确诊总人数的19.2%且乳腺癌确诊率呈现逐年上升趋势 [1] [2] 。根据癌细胞内蛋白分子的不同,乳腺癌可以分为雌激素受体、孕激素受体、人表皮生长因子-2三类,其中,约70%的乳腺癌患者表现为雌激素受体α (Estrogen Receptorα, ERα)阳性 [3] [4] 。雌激素受体α是一种转录因子核受体,其活性主要通过与雌激素结合来调控,该受体的活性受到雌激素的影响,研究发现该受体长期与雌激素结合是乳腺癌产生的因素之一 [5] 。
目前,对于ERα表达的乳腺癌患者的常规治疗是采用抗激素疗法,主要是通过限制雌激素受体的活性,从而达到控制体内激素水平的目的。抑制ERα成为了治疗乳腺癌的重要手段,因此在选择治疗乳腺癌的临床药物上,能够拮抗ERα活性的化合物成为了首选。当下,在药物研发过程中,建立化合物预测模型成为了筛选化合物活性的主要方法。另外,化合物具备良好的药代动力学性质和安全性,合称为ADMET性质,只有具备良好生物活性和ADMET性质的化合物,才能成为候选药物。
本文旨在根据提供的ERα拮抗剂信息,通过机器学习方法构建化合物生物活性的定量预测模型和ADMET性质的分类预测模型,从而达到为优化ERα拮抗剂的生物活性定量预测和ADMET性质分类预测提供服务的目的。
2. 化合物对ERα生物活性的定量预测模型
2.1. 分子描述符筛选
本文采用了斯皮尔曼相关系数法评价化合物各分子描述符对生物活性的相关程度,以找出要求的前20个影响最为显著的分子描述符;同时使用基于BP神经网络的MIV平均影响值算法,将729个分子描述符数据作为输入,pIC50预测值作为输出,计算得到各分子描述符所代表的相关系数,找出前20个影响最为显著的分子描述符。
Spearman相关系数能够确切的表明两个变量之间相关的程度,所以化合物的分子描述符对其生物活性的显著影响可由相关系数表征。本文中化合物的分子描述符属于等级数据,独立变量X和依赖变量Y之间无明显的正态分布和线性关系,故Spearman相关系数方法是用来求解该问题的有效方法。
针对化合物的分子描述符,Spearman相关系数可简化为:
(1)
其中,
为变量之间的等级差,一个数的等级,就是将其所在的一列数按照从小到大排序后这个数所在的位置,可以证明: 位于−1和1之间。Spearman相关系数的计算采用的方式不是取值本身,而是采用取值的等级。
平均影响值(MIV:mean impact value)用在神经网络中评价变量的相关性,该方法能够反映出自变量对输出神经元的影响 [6] 。本文将MIV结合BP神经网络,先训练好网络再将一个输入减少10%和增加10% (其它输入保持不变),将两组数据都输入到网络中,查看该输入的落差会引起网络输出多少的落差。如果引起的落差很少,则说明该输入对输出影响很小,否则,则认为对输出影响较大。
BP神经网络主要由三个层次组成,主要包含输入层,隐含层,输出层。首先数据通过输入层进入网络中,在隐含层进行网络处理,在输出层得到最后的结果。当训练得到的输出层的结果与预期的结果相差较大时,此时数据在神经网络中反向传播,在该阶段中通过调整网络权值,使得最终的输出结果与给定的预期结果满足一定的条件。本文建立的BP神经网络模型结构含有729个输入、1层隐含层以及1个输出,用于分析化合物对ERα生物活性的影响。
输入输出层:将729个分子描述符数据作为输入,pIC50预测值作为输出。故输入层神经元个数n = 729,输出层神经元个数m = 1。
隐层:确定神经元的个数是网格设计过程中重要的环节,当神经元个数过多的时候,网络计算量大,计算冗余,当网络神经元个数较少的时候,会影响网络的稳定性,计算误差大,无法达到预期的效果。隐含层网格神经元的个数主要与问题的复杂度和输出层预测结果与预期结果的误差条件有直接的关系。本文出于保证精度的考虑,经反复调试,最终确定隐层神经元个数为9个。神经网络结构示意如图1所示。

Figure 1. Schematic diagram of the neural network structure based on MIV BP
图1. 基于MIV的BP的神经网络结构示意图
由Spearman相关系数得到的各个分子描述符对化合物生物活性的相关系数如图2所示。
由MIV计算得到的各个分子描述符对化合物生物活性的相关系数如图3所示。

Figure 2. Schematic diagram of the distribution of the coefficients of correlation for calculating the molecular descriptor Spearman
图2. 计算分子描述符Spearman相关系数分布示意图
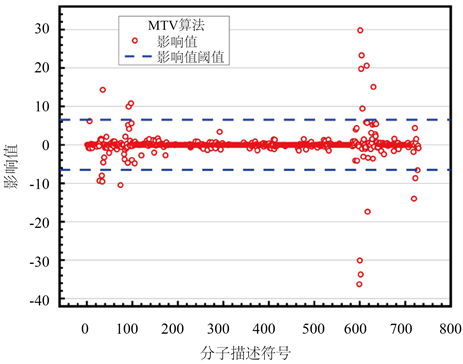
Figure 3. Schematic diagram of the effect of molecular descriptors on biological activity values for MIV calculations
图3. MIV计算分子描述符对生物活性影响值示意图
图中蓝色划线表示相关度阈值,对比spearman相关系数与MIV平均影响值算法得出的结果,MIV计算分子描述符对生物活性影响值大多数都集中在0附近,说明大多数分子描述符对生物活性值影响不大,只有少部分对生物活性影响较大;spearman相关系数计算得出的每一个分子描述符对生物活性影响较为离散,数据对比更加明显。本文认为相关系数的大小表征了分子描述符对生物活性的影响程度,因此选择对spearman相关系数计算出的结果进行排序,由此得到前20个对生物活性最具有显著影响的分子描述符及其相关系数,如表1所示。

Table 1. Significantly affects the table of correlation coefficients for molecular descriptors
表1. 较显著影响分子描述符相关系数表
2.2. 多元回归的预测模型
本文在选定好前20个对生物活性最具有显著影响的分子描述符的基础上,构建化合物对ERα生物活性的定量预测模型,通过模型对IC50值和对应的pIC50值进行预测。主要对50个化合物进行pIC50值预测,进而得到对应的50个IC50的预测值。
2.2.1. 数据处理
本文在建模中为了后续方便观察,先单独提取出这20个分子描述符所在的组对应的所有数据。由于这些数据都是化合物对ERα的生物活性采集的真实数据样本,所以这里不用对数据进行清洗,只需根据数据特征和后续各个模型的数据输入格式进行适当的分析和预处理即可,在此基础上对数据做了以下两个方面的处理:
1) 建立IC50与pIC50的负对数关系函数
本文已知IC50与pIC50的值成负对数关系,在现有的1974组IC50与对应的pIC50数值条件下,建立出IC50与pIC50的负对数关系函数,具体函数解析式如下所示:
(2)
式中:x为IC50;y为pIC50。
2) 归一化处理
由于得到的二十组分子描述符数据之间、各组数据与pIC50数据的量纲以及数量级可能存在差异,这种差异可能会对模型结果造成影响,为消除数据之间的影响,本文将对所有输入进行归一化处理。当原始数据经过数据标准化处理之后,经过归一化处理之后数据的指标量级相同,对于这种量级相同的数据适合进行综合对比评价,同时还可以提高模型的预测精度 [7] 。归一化目前有两种方法,一种是把数据修正为(0, 1)之间的小数,另一种是把有量纲表达式修正为无量纲表达式。本文将采用(0, 1)标准化方法,其计算公式如下:
(3)
式中:x为变量的值,min为该类变量中的最小值,max为该类变量中的最大值,
表示归一化的值。
2.2.2. 多元回归预测模型构建
研究一个因变量与两个或者两个以上的自变量的回归称为多元回归,多元回归主要在于反映一种现象或事物的数量,能够随着多种现象或者事物的数量的变动而发生相应变动的现象 [8] 。本文中由于分子描述符变量与pIC50值的线性或非线性关系未知,所以分别建立四个多元回归模型,四个模型为两个多元线性回归模型以及两个多元非线性回归模型。两个线性回归模型分别为原始数据线性回归以及稳健回归(使用加权最小二乘);两个非线性回归模型分别为最高项为二次多元非线性回归以及最高项为三次多元非线性回归。
多元回归预测模型程序最终选用Matlab软件进行编写,本文将数据按照分子描述符随机打乱,选取前80%作为训练集,后20%作为测试集。将训练集数据带入运行程序可得四个多元回归模型,其中两个线性回归模型中二十个分子描述符与pIC50值的回归关系式如下所示,
(4)
(5)
本文得出的四个回归模型如图4所示。
2.3. 回归模型验证
将测试集导入回归预测模型中,将预测得到的结果与其真实值进行对比,结果如图5所示。
可见多元回归预测值与其对应的真实值相近,说明回归模型具有较好回归结果,可较为真实地反应pIC50预测值。
图6计算各样本的预测残差百分比,可见原始数据线性回归以及稳健回归模型预测pIC50值残差百分比最大为0.5%左右,最高项为二次的非线性回归模型预测pIC50值残差百分比最大为0.23%左右,其中三次非线性回归模型预测结果最为准确,样本中预测的误差百分比均控制在0.2%以内。
通过计算可以得出原始数据线性回归预测模型决定系数为0.524,稳健回归预测模型决定系数为0.562,最高项为二次的非线性回归预测模型决定系数为0.722,最高项为三次的非线性回归预测模型决定系数为0.958。在多元回归预测模型中,最高项为三次的多元非线性回归预测模型最为准确。
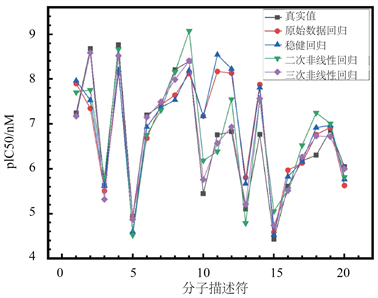
Figure 5. The prediction results of the four models of pIC50 are compared with the true values
图5. pIC50四个模型预测结果与真实值对比
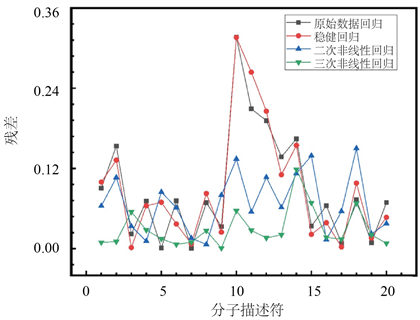
Figure 6. The four models of pIC50 predict the percentage of residual of the result
图6. pIC50四个模型预测结果残差百分比
3. 分类预测模型
本文使用729个分子描述符变量数据训练出来的模型,构建五种(Caco-2、CYP3A4、hERG、HOB、MN)分类预测模型,以实现对50种化合物的ADMET性质的预测。由于可以采用0、1两种形式来描述一种化合物的优劣两种属性,所以可简化为一个建立0/1分类预测模型的问题。难点在于涉及的变量及变量数据较多,不仅对有729个自变量,并且每组自变量有对应的1974个数据。筛选数据训练模型使其能够对应变量进行准确的0/1预测,针对这一难点,普通的求极值方法往往会陷入得到的结果是局部最优解的困境。为解决该问题本文采用广义回归神经网络(GRNN)和随机森林算法,利用数据训练模型,对比准确度选择最优模型对化合物进行相应的预测。
3.1. 广义回归神经网络(GRNN)与随机森林分类预测模型
广义回归神经网络(GRNN)是基于非线性回归理论的前馈式神经网络模型,作为径向基函数(RBF)网络的一种,是通过概率密度函数进行预测,其神经网络结构与BP神经网络结构相似,分为输入层、模式层、求和层和输出层 [9] 。
随机森林由多个决策树构成,决策树彼此之间互不关联。在进行分类预测的时候,每一棵决策树都要参与分类和判断,每一棵决策树都会产生独属于自己的分类结果,随机森林的最终结果是由决策树中分类最多的结果决定的 [10] [11] 。
3.2. 分类预测模型模型验证
本文将数据按照分子描述符随机打乱,选取前80%作为训练集,后20%作为测试集。利用训练集对模型调试,对测试集样本进行求解,选择最优模型对化合物ADMET性质进行相应的预测。
将预测模型分别带入测试集进行模型验证,预测得到的结果与其真实值进行对比计算正确率,首先测试Caco-2分类预测模型,结果如图7,图8所示。
GRNN分类预测模型预测正确率达到94.59%;随机森林预测模型预测正确率为81.89%。下面按照同样的方法对CYP3A4、hERG、HOB、MN分类预测模型的预测结果进行正确率计算,计算结果如表2所示。

Figure 7. The GRNN model calculates the accuracy of the prediction results of the test set Caco-2
图7. GRNN模型计算测试集Caco-2预测结果正确率

Figure 8. The random forest model calculates the accuracy of the prediction results of the test set Caco-2
图8. 随机森林模型计算测试集Caco-2预测结果正确率
Table 2. Comparison of the accuracy of prediction results
表2. 预测结果正确率的对比
可见通过GRNN预测模型预测对(Caco-2、CYP3A4、hERG、HOB、MN)的预测都能达到93%以上,随机森林预测正确率相对较低,可见在实际研究过程中利用GRNN预测模型能够有效预测所给50种化合物的ADMET性质。
4. 结论
本文充分利用非线性相关分析、多元回归分析、GRNN神经网络模型、随机森林算法等数据挖掘技术以及机器学习技术,建立了相关化合物定量结构-ERα活性以及定量结构-ADMET性质的定量预测模型。得出以下结论:
1) 利用Spearman相关系数计算方法得到729个分子描述符对生物活性影响程度,并对其进行排序,能得到影响程度排名前二十的变量。
2) 最高项为三次的多元非线性回归预测模型精确度最高,R方为0.958,残差百分比都在0.2%以内,使用此方法建立了相关化合物定量结构-ERα活性定量预测模型,能提高IC50值和对应的pIC50值预测的正确率。
3) 利用广义回归神经网络(GRNN)分类预测模型,该模型进行优化后预测正确率均在93%以上,使用此模型建立了相关化合物定量结构-ADMET性质的定量预测模型,提高预测的准确性。
参考文献