1. 引言
近年来,实验工作者发现在低温下用金属氢化物(CaH2, LiH, NaH)还原过渡金属氧化物能生成更高浓度氧缺陷(或者更低价态的金属离子),从而得到具有更少配位数的结构。2007年,Tsujimoto组 [1] 用CaH2还原SrFeO3得到了一种新型化合物SrFeO2。这种新型化合物和未掺杂的铜基高温超导母体材料SrCuO2 [2] 具有相同的结构,都属于层状结构化合物。尽管SrFeO2具有二维方形平面结构,但是它的尼尔温度(TN = 473 K)却比具有三维结构的氧化亚铁FeO(~200 K)要高许多。针对这一特性,2008年,Xiang等人 [3] 利用第一性原理计算方法分析SrFeO2的电子态密度以及能带,得出SrFeO2的基态磁构型为G类反铁磁序,作者认为SrFeO2的高尼尔温度是由于层间自旋交换作用引起的。另外,Pruneda 等人发现Fe2+离子中4s和 轨道的杂化对Fe2+中d轨道分布有着很大影响 [4] 。2009年,Ju等人发现SrFeO2具有显著的光学各向异性,预示了SrFeO2具有潜在的应用前景 [5] [6] 。与此同时,为了验证SrFeO2的结构稳定性,实验研究者进一步研究了掺杂和高压对SrFeO2结构以及磁性质的影响。实验发现SrFeO2具有很强的结构稳定性,可达到较高的Sr位掺杂浓度(100%的Ca掺杂 [7] ,30%的Ba掺杂 [8] )。对于Fe位掺杂,Co和Mn掺杂浓度均可至30% [9] ,Co掺杂对体系的G类反铁磁序的影响较小,而Mn的掺入使体系从G类反铁磁序变为顺磁性。已有的研究中,同价Sr位掺杂和Fe位掺杂,均没有引起体系输运性质的改变。而在高压下SrFeO2中FeO2层表现出半金属性,并且伴随着由反铁磁序到铁磁序的转变 [10] 。鉴于此,本文基于密度泛函理论,利用GGA + U的计算方法系统研究了模拟载流子严格定量掺杂以及A位不等价掺杂(La3+、K+)下,对SrFeO2磁电性质的影响。
轨道的杂化对Fe2+中d轨道分布有着很大影响 [4] 。2009年,Ju等人发现SrFeO2具有显著的光学各向异性,预示了SrFeO2具有潜在的应用前景 [5] [6] 。与此同时,为了验证SrFeO2的结构稳定性,实验研究者进一步研究了掺杂和高压对SrFeO2结构以及磁性质的影响。实验发现SrFeO2具有很强的结构稳定性,可达到较高的Sr位掺杂浓度(100%的Ca掺杂 [7] ,30%的Ba掺杂 [8] )。对于Fe位掺杂,Co和Mn掺杂浓度均可至30% [9] ,Co掺杂对体系的G类反铁磁序的影响较小,而Mn的掺入使体系从G类反铁磁序变为顺磁性。已有的研究中,同价Sr位掺杂和Fe位掺杂,均没有引起体系输运性质的改变。而在高压下SrFeO2中FeO2层表现出半金属性,并且伴随着由反铁磁序到铁磁序的转变 [10] 。鉴于此,本文基于密度泛函理论,利用GGA + U的计算方法系统研究了模拟载流子严格定量掺杂以及A位不等价掺杂(La3+、K+)下,对SrFeO2磁电性质的影响。
2. 计算方法
我们的计算基于VASP软件包 [11] - [14] ,采用全势缀加平面波方法(PAW)来描述电子-离子间相互作用。选用GGA + U (U = 6 eV)的方法处理SrFeO2体系中Fe的3d壳层的电子间关联效应。在PAW势中我们选用Sr中4s24p65s210个电子为价电子,Fe的3p63d64s2为价电子,O的2s22p4为价电子。利用Monhkorst-Pack方法产生K点,K点网格大小为10 × 10 × 10。
3. 计算结果与讨论
SrFeO2的晶体结构具有P4/mmm空间对称性,如图1所示。为了描述各种磁构型,平面内晶胞取 ,平面间晶胞取2c。每个晶胞由四个原胞组成,共包含16颗原子。图中绿色小球代表铁原子,蓝色小球代表锶原子,粉色小球代表氧原子。表1给出了优化后的晶格常数和体积,与实验值 [1] 相比十分接近。
,平面间晶胞取2c。每个晶胞由四个原胞组成,共包含16颗原子。图中绿色小球代表铁原子,蓝色小球代表锶原子,粉色小球代表氧原子。表1给出了优化后的晶格常数和体积,与实验值 [1] 相比十分接近。
图2给出了载流子掺杂SrFeO2的磁相图,掺杂范围从−0.25 e/f.u.到0.25 e/f.u.,这里负号表示空穴掺杂。可以看到,电子掺杂后体系的磁基态始终保持G类反铁磁序。而对于空穴掺杂,当掺杂浓度达到0.075 e/f.u.时,体系的磁基态发生了转变,由G类反铁磁态转变为A类反铁磁态。
进一步,我们计算了SrFeO2中自旋交换作用参数J1、J2和J3,见图1。其中J1是平面间最近邻铁原子之间的自旋交换作用,J2是平面内最近邻铁原子间的自旋交换作用,J3是平面间次近邻铁原子之间的自旋交换作用。
对于电子掺杂,可以看到平面内最近邻交换参数J2 (即JFe-O-Fe)几乎不受电子掺杂的影响,其值基本保持不变。平面间最近邻交换参数J1 (即JFe-Fe)仍为负值,其绝对值略微减小,表明层间反铁磁耦合作用随着电子掺杂浓度的增加而稍有减弱。
对于空穴掺杂,我们发现平面内最近邻J2随空穴掺杂的变化最为明显。当空穴掺杂浓度达到磁相变的临界浓度(−0.075 e/f.u.)时,J2由负值变为正值;并且当空穴掺杂浓度达到−0.25 e/f.u.时,J2增大到8.1 meV,这表明层内铁原子之间的铁磁耦合随着空穴浓度的增大而逐渐增强。值得一提的是,在高空穴浓度下层间最近邻J1由负值变为正值(0.25 e/f.u.时为1.6 meV),这似乎与A类反铁磁基态不相符合,但我们注意到层间次近邻J3在高空穴掺杂浓度下由正值变为负值,并且由于J3的数目要比J1的数目多,它们相互竞争使体系的磁基态在高空穴掺杂浓度下表现为A类反铁磁序是可以理解的。
为了弄清背后的物理机制,我们在图3中给出了掺杂后体系的总态密度。与块材相比,掺入电子后,电子态密度形状并几乎没有发生改变,只是整体向能量更低的位置偏移。而对于空穴掺杂,当空穴掺杂浓度小于临界浓度时(图中黑色实心线),体系未发生磁相变。相比于块材,总态密度基本特征几乎没有改变,只是整体稍微向右平移。当空穴浓度高于临界浓度时,体系发生磁相变,可以看到在费米能级附近,态密度明显展宽。
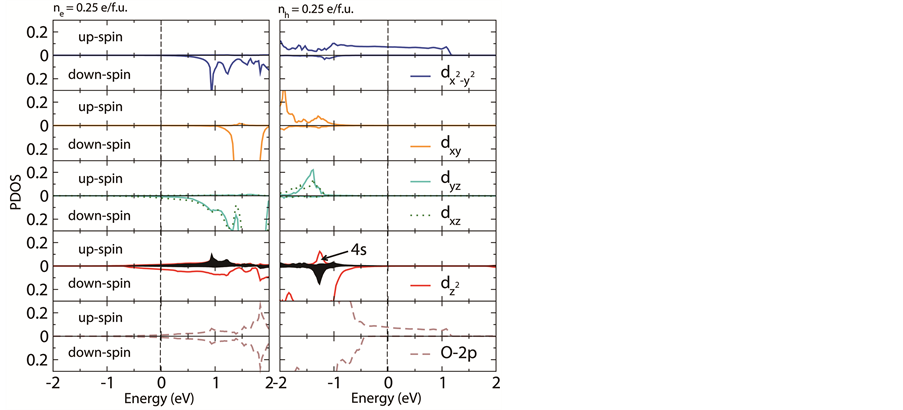
进一步,我们给出了SrFeO2高掺杂浓度下Fe中各3d轨道和O 2p轨道分波态密度。图4(a)可以看到电子掺杂下引入的额外电子主要占据在Fe dz2及与之强烈杂化的4s轨道上,也有一部分电子占据在简并的dxz、dyz自旋向下轨道上。随着电子掺杂浓度逐渐升高,dz2轨道逐渐被电子完全占据,从而导致dz2轨道上额外电子的占据对磁性影响较小。而dxz、dyz简并轨道对磁性交换的影响较大,随着电子掺杂浓度的增加,层间原有的反铁磁交换作用减弱,对应图2中层间最近邻交换作用J2的值减小,暗示了体系的磁性逐渐由三维变为二维。
对于空穴掺杂,见图4(b),我们发现引入的空穴主要占据在Fe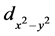 轨道以及与之强烈杂化的O 2p轨道。我们认为当相邻两个
轨道以及与之强烈杂化的O 2p轨道。我们认为当相邻两个 轨道以铁磁方式耦合,电子(也可以说是空穴)可以在Fe-O-Fe键中自由
轨道以铁磁方式耦合,电子(也可以说是空穴)可以在Fe-O-Fe键中自由
跃迁,从而使体系动能降低。因此,随着空穴掺杂浓度的增大,层内变为铁磁耦合。
最后,我们用La或K元素取代Sr来考察电子掺杂和空穴掺杂对体系的影响。优化后的晶格常数如表2所示。考虑不同磁构型时,计算得到的能量相对值如表3所示。可以发现La掺杂下体系依然保持G类反铁磁序,然而K掺杂后体系的磁基态变为A类反铁磁序。
图5(a)给出了(Sr0.75La0.25)FeO2的总态密度。可以看到La掺杂后体系的总态密度与电子掺杂浓度0.25 e/f.u.时相比,除了导带的形状有一定的改变以外(La的贡献),在费米能级附近态密度特征基本一致。与

Figure 1. Crystal structure of the SrFeO2 and magnetic exchange couplings
图1. SrFeO2的晶体结构图及磁性交换作用

Figure 2. The magnetic phase diagram and corresponding total energies and magnetic exchange coupling constants of SrFeO2 with carrier doping
图2. 载流子掺杂下SrFeO2磁相图和相应的总能及磁交换作用系数

Table 1. The calculated and experimental crystal structure parameters of SrFeO2
表1. SrFeO2块材晶格常数计算值和实验值对比

Figure 3. Total density of states of SrFeO2 under different (a) electron and (b) hole doping concentration for both spin up and spin down channels. The shaded area is for pristine SrFeO2
图3. (a) 电子和(b) 空穴掺杂浓度下体系的总态密度。阴影区域为未掺杂的SrFeO2
 (a) (b)
(a) (b)
Figure 4. Orbital resolved density of states at the (a) electron and (b) hole doping concentration of 0.25 e/f.u.
图4. 0.25 e/f.u. (a) 电子掺杂浓度和(b) 空穴掺杂浓度下Fe的3d轨道和O的2p轨道分波态密度
Table 2. Optimized structure parameters of (Sr0.75La0.25)FeO2 and (Sr0.75K0.25)FeO2
表2. (Sr0.75La0.25)FeO2和(Sr0.75K0.25)FeO2的优化晶格常数
Table 3. The relative total energies of (Sr0.75La0.25)FeO2 and (Sr0.75K0.25)FeO2 with different magnetic ordering
表3. (Sr0.75La0.25)FeO2和(Sr0.75K0.25)FeO2不同磁构型下的相对总能量值
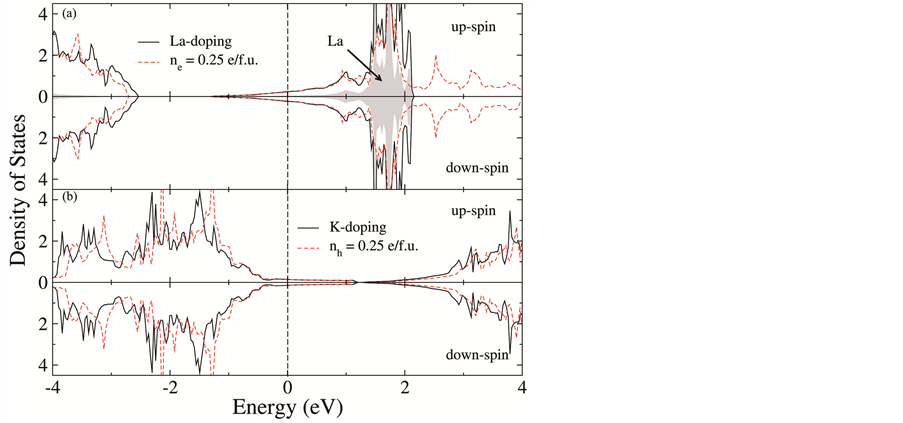
Figure 5. The total density of states of (a) (Sr0.75La0.25)FeO2, (b) (Sr0.75K0.25)FeO2
图5. (a) (Sr0.75La0.25)FeO2的总态密度和(b) (Sr0.75K0.25)FeO2的总态密度

Figure 6. The partial density of states for Fe 3d/4s orbitals and O 2p orbitals of (a) (Sr0.75La0.25)FeO2 and (b) (Sr0.75K0.25)FeO2
图6. (a) (Sr0.75La0.25)FeO2和(b) (Sr0.75K0.25)FeO2中Fe 3d/4s轨道与O 2p轨道态密度
之类似,图5(b)中(Sr0.75K0.25)FeO2费米能级附近态密度形状也与空穴掺杂−0.25 e/f.u.时的态密度基本一致。从图6可以明显看出K元素掺杂时Fe 轨道被部分占据。
轨道被部分占据。
4. 总结
利用第一性原理计算方法,我们探究了载流子掺杂对SrFeO2电子结构和磁性质的影响。我们发现电子掺杂后体系的磁基态依然保持G类反铁磁序,而对于空穴掺杂,当达到临界浓度(0.075 e/f.u.)后,体系的磁基态转变为A类反铁磁序。我们发现这是由于Fe离子的 轨道部分占据驱动了层内Fe离子之间的铁磁耦合。最后,我们分别用La和K对Sr位进行替换,也得到了同样的结论。
轨道部分占据驱动了层内Fe离子之间的铁磁耦合。最后,我们分别用La和K对Sr位进行替换,也得到了同样的结论。
*通讯作者。