1. 引言
核小体作为真核生物染色质高级结构的基本单位,是由DNA与组蛋白结合而成的典型生物大分子,由约147碱基对的DNA分子盘绕组蛋白八聚体上形成的核心DNA序列与长度约10~50碱基对的连接序列两部分组成 [1] [2] [3] 。组蛋白八聚体是由高度保守的H2A,H2B,H3和H4各二分体组成,在组蛋白H1的连接作用下,形成一个高级分子结构 [2] 。核小体的特殊结构限制了负责基本生命过程的蛋白质与围绕组蛋白上的DNA接触,所以在基因表达过程中它的形成以及在染色质上的精确定位在基因表达过程中起着无可替代的作用,直接或间接地影响转录等基本生物过程 [3] [4] [5] 。
本研究利用实验检测的高分辨率的人类CD4 + T细胞中休眠状态下核小体占据率数据,分析和比较了人类基因组核小体定位与缺乏序列的一些特征。
2. 材料与方法
2.1. 材料
人类CD4 + T细胞全基因组的核小体占据数据来自于Schones等 [6] 所做的工作。数据通过MNasese方法获得。该数据为人类CD4 + T细胞在休眠状态下和被CD3抗原激活后的全基因组核小体占据bed数据。网址为http://dir.Nhlbi.nih.gov/papers/lmi/epigenomes/hgtcellnucleosomes.aspx。
人类全基因组序列数据来源于UCSC基因组数据库hg18版本http://hgdownload.cse.ucsc.edu/。
本工作中数据集构建包括核小体占据序列数据集(简写为nucleosome)与核小体缺乏序列数据集(简写为null)两部分。核小体占据序列数据集即为基因组上DNA被核心组蛋白占据的部分。首先,扫描基因组上碱基被目标探针覆盖的次数,经过统计占据率分布后选取分数13为阈值进行初步筛选,即将基因组上被探针覆盖13次及13次以上的区域作为定位备选集。而后,进行长度筛选,取连续覆盖范围201 bp的序列。最后,利用blast软件进行序列比对,将完全重复或高度相似的序列进行归类,每类中只保留其中一条序列,最终筛选得到核小体占据序列36,777条。缺乏序列数据集为核小体缺乏DNA序列数据集,它的构建与定位序列数据集的构建的相似。首先在基因组上进行探针覆盖次数扫描,只选取0覆盖的区域作为缺乏备选集(为了保证可靠性,覆盖次数在0与13之间的被认为是模糊定位区域而放弃不用)。在进行序列长度筛选时,选用连续0覆盖范围在101 bp的序列来构建缺乏区数据,经过挑选后获得332,772条长度为101 bp的序列。为了避免占据、缺乏数据集间因序列长度造成的统计偏差,又将定位区每条序列拆成1~101 bp,101~201 bp两部分。
2.2. 方法
2.2.1. K-Mer出现频率
如果把长度为k的核甘酸片段看作是一种“字”(k-mer),那么k-mer的频数就是长度为
的窗口在核苷酸序列上顺次移动时出现的次数,即“字”在序列上出现的次数。当k较大时,k-mer的频数分布构成了基因组的一个“等价表示”,即k-mer的频数分布可以唯一地确定基因组序列 [7] [8] 。组成DNA序列的核苷酸有4种:A (腺嘌呤),G (鸟嘌呤),T (胸腺嘧啶)和C (胞嘧啶),k-mer共有4k种。对于序列总长度为LE的DNA序列k-mer出现频率定义为Pi。
(2.2.1)
其中Ni为第i个k-mer在序列中出现的频数,
。
2.2.2. GC含量、CpG相对丰度
G + C含量(GC (content))是基因组结构中的一个重要的因素 [9] ,定义
(2.2.2)
Kariln等人通过对基因中寡聚核苷酸的相对丰度的研究,发展出一套用数值比较物种间不同基因以及物种基因的不同部位的方法 [10] 。相对丰度的思想认为由于DNA序列中碱基相邻的频率并不是独立的,也就是说碱基的分布不是随机产生,相邻碱基的频率不等于单个碱基频率的乘积。CpG相对丰度(ρCG)描述CpG二核苷酸的实际频率与从其组成核苷酸的频率估算的理论频率的差别,其定义如下:
(2.2.3)
2.2.3. K-Mer频数分布率
为了反映各k-mer在序列中出现的频数的分布状况,我们首先分别计算了每条序列中各k-mer出现的频数,统计每个k-mer在不同频数区间出现的条数,则一定区间出现的各k-mer的条数率定义为k-mer频数分布率FAj:
(2.2.4)
Lij为第j个k-mer出现在第i个频数区间的序列条数,分母为该数据集序列总条数。
2.2.4. 二核苷酸位置频率
对于核小体定位序列数据集建立二核苷酸位置频率SAj:
(2.2.5)
Lij为第i个2-mer出现在第j个位点上的序列条数,分母为该数据集序列总条数。
3. 结果与讨论
3.1. 核小体定位序列偏好C、G
根据(2.2.1)、(2.2.2)式我们对核小体定位序列数据集、核小体缺乏序列数据集的序列总数、碱基频率、2-mer频率以及GC含量做了统计,结果见表1和表2。

Table 1. The frequency of single-base, GC content and CpG relative abundance in nucleosome and null sequences
表1. 核小体定位、缺乏序列单碱基频率、GC含量、CpG相对丰度数据统计
Table 2. The frequency of dinucleotides in nucleosome and null sequences
表2. 核小体定位、缺乏序列2-mer出现频率统计
统计结果显示核小体定位序列单核苷酸G、C的含量明显高于A、T。A + T含量越高,序列的刚性越强,越不利于DNA的弯曲。因此,定位序列的单核苷酸A和T的含量低有助于核小体DNA缠绕组蛋白八聚体。通过计算亦证实定位序列的整体G + C含量(G + C = 0.5008)显著高于缺乏序列区(G + C = 0.4093)。核小体定位序列中出现频率最高的八个二联体依次为TG、CA、AG、CT、GG、CC、AA、TT;缺乏序列中出现频率最高的八个二联体依次为TT、AA、AT、TG、CA、CT、AG、TA。定位序列中出现频数最高的八个4-mer依次为CAGG、CCTG、CTGG、CCAG、GCAG、TGTG、CACA、GCTG;缺乏序列中出现频数最高的八个四联体依次为TTTT、AAAA、AAAT、ATTT、TTTA、TAAA、ATAT、AATT。缺乏序列高频次出现的4-mer由碱基A和T组成。核小体定位序列中GC含量显著高于AT含量而高频率的k-mer并不完全是只包含G、C碱基。
二核苷频率AA、TT在两类序列的频率差别显著(图1)。图2显示CG二核苷在两类数据集出现的频率都最小,定位区仅为0.017,缺乏区占0.0199,均低于随机水平0.0625,CG的低频现象也与基因组中CG缺乏的现象吻合。
3.2. k-mer频数分布率特征
利用2.2.3方法得GC含量及各单碱基频数分布率,由图3显示核小体定位序列数据集中GC含量变化范围是在5到85之间,其中含量在35到70之间的序列占据了序列总数的98%。核小体缺乏序列的GC含量多数集中在0到65之间,多数缺乏序列的GC含量低于定位序列。单碱基的频数分布在两类序列集中没有显著区别(图4)。
3.3. 二核苷酸位置频率特征
我们利用长度是201 bp的核小体定位序列数据集计算每个二核苷酸出现在各位点的概率作图5,发现在相同位置不同二核苷酸出现的频率不同,不同位置相同二核苷酸出现的频率也不同。表3列出了16种二核苷酸在核小体定位序列中的平均占据率。假设核小体定位序列不存在统计特性,那么二核苷酸位置频率的值在任意位置应该是随机产生的,在1/16大小浮动。而且表3可以看出所有二核苷酸在定位序列中并没有平均分配,这表明定位序列中二核苷酸位置频率存在一定统计特性。
4. 结论
分析核小体定位序列统计特征发现人类核小体定位序列与缺乏序列在GC含量和k-mer分布存在显著差异。核小体定位序列偏好单碱基G + C,偏好二核苷CC、CG、GG、GC;核小体缺乏序列偏好单碱

Figure 1. The frequency of single-base in nucleosome and null sequences
图1. 核小体定位、缺乏序列单碱基出现频率
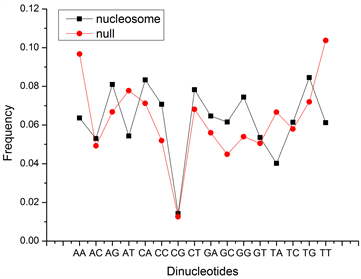
Figure 2. The frequency of dinucleotides in nucleosome and null sequences
图2. 核小体定位、缺乏序列2-mer出现频率

Figure 3. The frequency of distribution on GC content in nucleosome and null sequence
图3. GC含量在核小体定位缺乏序列分布率
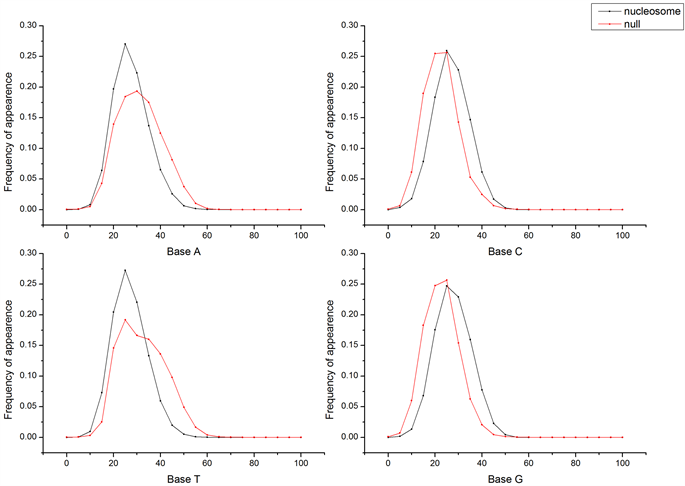
Figure 4. The frequency of distribution on single-base in nucleosome and null sequence
图4. 单碱基在核小体定位、缺乏序列分布率
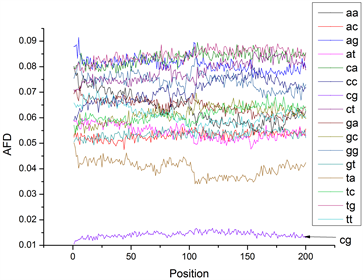
Figure 5. Nucleosome location sequence dinucleotide location frequency
图5. 核小体定位序列二核苷酸位置频率
Table 3. Average occupancy rate of 16 dinucleotides in nucleosome localization
表3. 16种二核苷酸在核小体定位中的平均占有率
基A + T,偏好二核苷TT、AA、AT、TA,表明与C和G相关的二核苷、甚至k核苷(k > 2)在核小体形成及其功能行使上以基本的功能单位参与作用;16种二核苷在核小体定位序列和核小体缺乏序列上的平均占有率同样存在上述偏好性,这不仅支持了与C和G相关的二核苷、甚至k核苷(k > 2)作为基本功能单位的观点,同样表明此类二核苷及k核苷对基因转录及进化同样具有深刻的意义。本文只是从k核苷偏好性及其组成偏好性出发进行了分析,进一步研究将对此类探索具有重要意义。
基金项目
内蒙古工业大学重点研究项目(ZD201614)。
参考文献