摘要:
本文采用密度泛函理论的方法在B3LYP/6-31G(d)水平上对精氨酸进行了研究,计算得到了精氨酸的稳定分子构型及其红外振动光谱,研究发现,根据分子振动类型的不同可将红外光谱划分为四个区域,其中在(0~800) cm−1区域对应的分子的振动类型主要为各基团的整体转动或摆动;在(800~1500) cm−1范围内谱峰所对应的分子振动模式主要是面外弯曲振动,在(1500~2800) cm−1分子的振动类型主要是面内弯曲振动,在(2800~4000) cm−1区域,分子的振动类型主要为各原子的伸缩振动。此外,在光谱中出现了无红外活性和简并的现象。
Abstract:
In this paper, the arginine is
studied at the 6-31G(d) basis set level, using the B3LYP method of density
functional theory. Through calculation, the stable structure and its infrared
spectrum are gained. The results show that the infrared vibration spectrum
includes four regions on the base of molecular vibration type. The vibration
type is the whole rotation or oscillation of group in the region of (0 - 800) cm−1.
In the region of (800 - 1500) cm−1, the vibration type is main
out-plane bending vibration, the out-plane bending vibration mode is the main
vibration type in the region of (1500 - 2800) cm−1, and in the
region of (2800 - 4000) cm−1, the vibration type is main stretching
vibration mode. Besides, there are non-infrared and activity in the infra-red
spectrum.
1. 引言
精氨酸Arginine是构成蛋白质的基本单元,是组成人体蛋白质的21种氨基酸之一。天然精氨酸为L-型,从水中结晶的产物含两分子结晶水,在乙醇中结晶是无水物。性状为白色斜方晶系晶体或白色结晶粉末,熔点244℃,经水重新结晶后,于己于105℃失去结晶水。其水溶液呈强碱性,可以从空气中吸收二氧化碳。溶于水(15%, 21℃),不溶于乙醚,微溶于乙醇 [1] 。天然品大量存在于鱼精蛋白中。研究表明,精氨酸对治疗心血管疾病、高氨血症、肝脏机能障碍等疾病颇有效果。精氨酸是一种双基氨基酸,对于成人来说虽然不是必须氨基酸,但在有些情况如机体发育不成熟或在严重应激条件下,如果缺乏氨基酸,机体便不能维持正氮平衡与正常的生理功能。若缺乏精氨酸会导致血氨过高,甚至昏迷。婴儿若先天性缺乏尿素循环的某些酶,精氨酸对其也是必须的,否则不能维持其正常的生长与发育。精氨酸的重要代谢功能是促进伤口的愈合作用,它可促进胶原组织的合成,故能修复伤口。
密度泛函方法 [2] [3] 是在量子力学基础上发展起来的一种计算方法,已被广泛地应用于计算化学、物理等领域,可成功地预测和解释各种材料中存在的现象。调研发现,目前国内外对精氨酸的理论和实验研究较少。本文运用Gaussian 09计算程序,在B3LYP/6-31G(d)水平上利用量化计算的方法对精氨酸分子的结构特点和红外光谱分布及形成规律进行了研究,以期能为精氨酸的快速检测提供理论支持。
2. 计算方法
首先利用Gauss View和Chem Office软件猜测构建了精氨酸分子的初始构型,运用Gaussian09计算程序中的HF/3-21G(d)方法对分子构型进行了初步优化,然后利用B3LYP/6-31G(d)方法,在优化所得初始构型基础上优化得到了分子和离子的最终稳定构型,最后对所得构型进行频率计算和分析,得到了分子和离子的红外振动光谱。文中频率计算采用0.9613 [4] 的修正因子进行矫正。
3. 结果与讨论
3.1. 精氨酸分子的结构
精氨酸分子结构如图1所示。表1中列出了精氨酸分子的部分结构参数。分子结构中骨架C、N、O原子呈现出链式结构的特点。结构中共存在N-H、N-C、C-H、C-C、C=O、C-O、O-N、O-H共6种化学键。在分子结构中,3C-6C键和9C-6C键键长近似相等,均为1.533 Å,9C-12C键键长为1.556 Å,12C-14C键键长为1.530 Å;结构中15N-1C键和18N-1C键键长均为1.466 Å,而21N-1C键键长为1.452 Å,23N-12C键键长为1.459 Å;14C=26O键键长为1.212Å,14C-27O键键长为1.356Å;分子中15N-16H和15N-17H分子键键长均为1.021 Å,15N-19H、21N-22H和18N-22H三个分子键键长均为1.019 Å,23N-24H和23N-25H两个分子键键长均为1.023 Å;结构中12C-13H、10H-9C、3C-4H键键长均为1.097 Å,11H-9C、5H-3C和2H-1C三个键的键长均为1.101 Å,7H-6C键长为1.099 Å,8H和6C键长为1.094 Å。结构中各原子间所成键角中,∠6C-3C-1C、∠28H-27C-14C和∠22H-21N-1C均为106˚,∠17H-15N-1C、∠18N-1C-15N和∠23N-12C-9C均为111˚,∠7H-6C-3C、8H-6C-3C、∠10H-9C-6C、∠11H-9C-6C和∠20H-18N-1C均为110˚,∠13H-12C-9C、∠16H-15N-1C和∠24H-23N-12C均为108˚,∠26O-14C-12C为125˚,∠4H-3C-1C为136˚,∠5H-3C-1C和3C-1C-2H均为89˚。在各原子所形成的二面角中,∠12C-9C-6C-3C和∠28H-27C-14C-12C两个二面角均为180˚,因此分别参与形成三个二面角的四个原子处于同一平面内。

Figure 1. Stable structure of arginine
图1. 精氨酸分子的稳定构型

Table 1. Partial structural parameter of arginine
表1. 精氨酸分子的部分结构参数
3.2. 精氨酸分子的红外振动光谱
物质因受红外光的作用,引起分子或原子基团的振动,若振动频率恰与红外光波段的某一频率相等时就引起共振吸收,使光的透射强度减弱,由此所得的光谱称为红外光谱 [5] 。红外吸收强度决定于振动时偶极矩变化的大小。若振动过程中偶极矩变化较大,则跃迁几率就会越大,红外光谱强度就会较强;反之,强度就会较弱 [6] [7] 。
标准状况下,精氨酸分子的红外光谱如图2所示。为了更好地分析精氨酸的特性,我们对精氨酸分子的红外光谱进行了详细分析,对光谱中出现的各条谱线进行了指认,对谱线形成所对应的各振动形式进行了归属。分析发现,精氨酸分子的红外光谱中谱线数目少于分子的简正振动的数目,分析认为这主要是由于光谱中出现了无红外活性的现象 [8] 。根据分子的振动类型的不同可将精氨酸分子的红外振动光谱划分为四个区域:(0~800) cm−1、(800~1500) cm−1、(1500~2800) cm−1和(2800~4000) cm−1。此外,基本上红外光谱的每条谱峰都是由多个振动模式叠加而成的,且在光谱中出现了无红外活性和简并的现象。
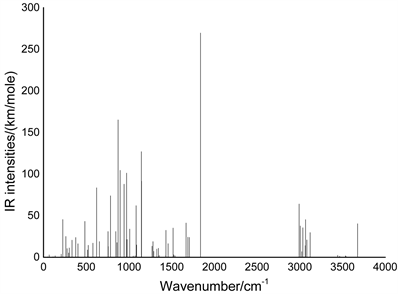
Figure 2. Infra-red spectrum of arginine
图2. 精氨酸分子的红外光谱
在(0~800) cm−1区域,分子振动类型主要是各基团的整体转动或摆动。本区域的红外光谱强度相比其他区域的光谱强调相比强度整体较低,且在该区域中出现了无红外活性的现象,这主要是由于在该区域中的谱峰所对应的分子振动没有在较大程度上引起分子偶极矩的变化造或有些分子振动没有引起偶极矩的变化而引起地。此外还在多个频率位置上出现了简并现象。该区域中最强峰出现在83 cm−1位置,它是由含18N的NH2基团整体摆动引起地。次强峰出现在784 cm−1位置,它是由含15N的NH2和含23N的两个NH2基团的个子整体摆动的复合振动引起地。225cm-1位置谱峰是由含15N的NH2基团的左右摆动和汗23N的NH2基团的整体转动以及含18N的NH2基团的整体转动的复合振动引起地,而484 cm−1位置的振动峰则是由18N的NH2基团整体转动引起地。此外,在755 cm−1位置出现了三重简并现象,分别与含C18的NH2基团的整体转动、含C23的NH2基团的整体转动和18N的NH2基团整体摆动相对应,并且在112 cm−1、47 cm−1和20 cm−1三个位置出现了无红外活性的现象,其中112位置的分子振动为含N15的NH2基团的整体转动,47 cm−1位置的分子振动类型为含N23的NH2基团的整体转动的左右摆动,20 cm−1位置的分子振动类型为含23C的NH2的整体摆动和含N18的NH2基团的整体转动的复合振动。
在(800~1500) cm−1区域,分子的振动类型主要是面外弯曲振动,即面外摇摆振动和扭曲振动。该区域最强峰出现在872 cm−1位置,它也是整个光谱中的次强峰,是由16H和17H的面外摇摆振动引起地,次强峰出现在1145 cm−1位置,是由19H和20H的扭曲振动引起地,而897 cm−1位置的谱峰则是由16H和17H的扭曲振动与24H和25H的面外摇摆振动的复合振动引起地,而同时16H和17H的摇摆振动与24H和25H的面外摇摆振动的复合振动的在1148 cm−1位置引起一较强共振峰。此外,在该区域光谱中,出现了双重简并现象,位置在978 cm−1,分别与10H和11H的扭曲振动,以及24H和25H的扭曲振动相对应;同时,在1458 cm−1位置出现了三重简并现象,分别与16H和17H的扭曲振动与19H和20H的扭曲振动的额复合振动,25H和24H的面外摇摆振动,以及10H和11H的面外摇摆振动相对应。此外,由于8H和7H的剪式振动及19H和20H的面外摇摆振动时均没有引起偶极矩的变化,致使在1072 cm−1和1050 cm−1两个位置出现了无红外活性的现象。
在(1500~2800) cm−1区域,分子的振动类型主要为面内弯曲振动,即面内摇摆振动和剪式振动。该区域最强峰出现在1838 cm−1位置,它也是整个红外光谱的最强峰,是由19H和20H的剪式振动、24H和25H的面内摇摆振动及26O和27O的剪式振动的复合振动引起地。该区域的次强峰出现在1668 cm−1位置,起对应的分子振动类型是19H和20H的面内摇摆振动,如图3所示。16H和17H的剪式振动及7H和8H的面内摇摆振动的复合振动在1517位置引起一较强共振峰。16H和17H的剪式振动在1689 cm−1位置引起一较强共振峰,振动模式如图4所示。24H和25H的剪式振动在1708位置引起一共振峰,而7H和8H的剪式振动、4H和5H的剪式振动及22H的面内摇摆振动的复合振动在1525 cm−1位置引起一共振峰。此外,在1539 cm−1和1515 cm−1两个位置的共振峰出现了无红外活性的现象,它们分别与4H和5H的剪式振动及7H和8H剪式振动的复合振动,以及10H和11H的剪式振动及7H和8H剪式振动的复合振动相对应。
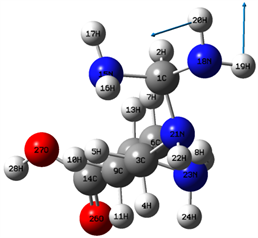
Figure 3. 1668 cm−1 vibration mode
图3. 1668 cm−1振动模式

Figure 4. 1689 cm−1 vibration mode
图4. 1689 cm−1振动模式
在(2800~4000) cm−1区域,分子的振动类型主要为各原子的伸缩振动。区域最强峰出现在2992 cm−1位置,主要是由2H的伸缩振动引起地,区域次强峰出现在3067 cm−1位置对应,它与4H的伸缩振动对应,而28H的伸缩振动在3676 cm−1位置引起一较强共振峰。在3006 cm−1位置的共振峰与4H和5H的对称伸缩振动相对应,而10H和11H的对称伸缩振动则与3021 cm−1位置共振峰相对应。7H的伸缩振动在3035 cm−1位置形城一较强共振峰,其强度与8H伸缩振动在3120 cm−1位置共振峰相近。在反对称伸缩振动引起地共振峰中较强的共振峰出现在3536 cm−1位置,它是由16H和17H的反对称伸缩振动形成地,而19H和20H的反对称伸缩振动,以及24H和25H的反对称伸缩振动在3558 cm−1和3490 cm−1两个位置所引起的两个共振峰出现了无红外活性的现象,其分子振动模式如图5和图6所示。
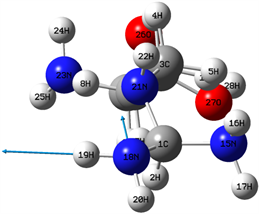
Figure 5. 3558 cm−1 vibration mode
图5. 3558 cm−1振动模式

Figure 6. 3490 cm−1 vibration mode
图6. 3490 cm−1振动模式
4. 结论
本文通过量化计算的方法获得了精氨酸分子的稳定构型和红外振动光谱,并对分子构型特点和红外光谱分布规律进行了研究分析。分析发现,精氨酸分子的红外光谱主要分布在四个区域,在(0~800) cm−1区域,谱线强度整体较低,振动类型以各基团的整体转动或摆动为主。在(800~1500) cm−1区域,分子的振动类型主要是面外弯曲振动,即面外摇摆振动和扭曲振动。在(1500~2800) cm−1区域,分子的振动类型主要为各原子面内弯曲振动,即面内摇摆振动和剪式振动。在(2800~4000) cm−1区域,分子的振动类型主要为各原子的伸缩振动。此外,红外光谱中出现无红外活性和多重简并的现象。