1. 引言
NO2是形成酸雨、光化学烟雾和雾霾的重要物质,对环境和人类健康有很大的危害 [1] [2]。因此,NO2的治理一直都是研究的热点之一。固体吸附法利用吸附剂对NO2发生吸附,因其脱除效果好、成本低、易于再生而被广泛应用。选择性催化还原法作为消除废气中NO2的主要方法,催化效率与固体表面上的吸附反应密切相关 [3]。因此,研究NO2在固体表面的吸附反应对于提高脱除效率至关重要。
近年来,钙钛矿型氧化物(ABO3)因其优良的热稳定性、化学稳定性、廉价以及易于改性等优点被广泛应用于光催化 [4]、化学催化 [5] [6] [7] 和气体传感器 [8] [9] [10] [11] [12]。研究表明,LaFeO3与许多气体分子能够发生强烈的吸附作用,如O2 [13]、CO [14]、CO2 [15]、HCHO [16]、NO [17]。这表明LaFeO3是优良的气体吸附剂。通过对NO2在Fe及FeS2表面吸附的理论计算发现,NO2都更加倾向于吸附在Fe原子上形成化学吸附。吸附反应主要基于N的2p轨道和Fe的3d轨道发生强烈杂化形成的Fe-N化学键 [18] [19],结果表明Fe原子是吸附反应的活性点位。因此,研究NO2在LaFeO3表面的吸附机理对于实际应用具有重要的意义。另外,在钙钛矿材料的应用研究中,利用离子掺杂优化材料的性能是研究的热点之一。钙钛矿(ABO3)中的A位离子一般为碱土金属或稀土金属,具有稳定钙钛矿材料结构、调节B位离子价态的作用。B位离子为过渡金属。实验研究表明,钙钛矿材料的活性主要取决于B位元素。Hernández等人研究了La0.6Sr0.4BO3 (B = Fe, Mn, Ti)对汽油燃烧产物炭黑的催化氧化,研究中发现改变B位离子能够影响结构的形态,电荷补偿机制及氧化还原性能 [20]。Baiker等人研究了ACoO3 (A = La, Pr, Nd, Gd)中A位离子对催化氧化甲烷的影响,实验结果表明,A位离子对提高材料的催化活性没有帮助。ACoO3在甲烷催化反应中的实际活性成分是Co3O4 [21]。此外,Sun等人研究了Ca2+,Sr2+和Ba2+的A位掺杂对LaFeO3吸附HCHO性能的影响,发现上述离子掺杂对LaFeO3吸附HCHO没有明显影响 [16]。因此,B位离子掺杂对钙钛矿的性能有更大影响。Kizaki等人研究了氧空位缺陷对LaFeO3体相中LaO(001)表面吸附NO的影响,结果表明,存在氧空位能大幅增强表面对NO的吸附作用 [22]。因此,研究B位离子掺杂及氧空位缺陷对LaFeO3吸附性能的影响是有必要的。
本文基于密度泛函理论(DFT)对NO2在LaFeO3 (010)表面可能的吸附点位进行了模拟计算,旨在通过分析吸附后的相关参数、电子状态等获得吸附反应机理及活性点位。同时,进一步探究了X2+ (X = Ni, Cu, Zn)掺杂对LaFeO3 (010)表面吸附NO2的影响。
2. 方法与模型
所有的计算都应用Materials Studio软件的CASTEP程序完成。交换关联能选择GGA-PBE方法描述 [20],同时考虑了自旋极化。价电子用平面波基组展开,截断能为400 eV。平面波的计算中采用超软赝势 [20] 来描述离子芯与价电子之间的相互作用。
LaFeO3在常温下为正交相结构,空间群为Pnma62,其磁性表现为G-型反铁磁态 [22]。(010)晶面为LaFeO3的主要解理面。立方相LaFeO3的晶格常数为a = 5.535Å、b = 7.888Å和c = 5.599Å。原子的分数坐标为Fe (0.0000, 0.0000, 0.0000)、O1 (0.2709, 0.0347, 0.2294)、O2 (0.4940, 0.25, 0.5644) 、La (0.5184, 0.25, 0.0089) [13]。由于Fe属于3d过渡金属,一般认为需要考虑电子强关联作用,因此采用GGA+U计算方法 [23]。LaFeO3体相计算采用4 × 3 × 4的Monkhorst-Pack k点网格完成布里渊区积分。LaFeO3 (010)表面的计算使用4 × 2 × 1的k点网格。几何优化和能量计算中,设置的能量收敛标准为2.0 × 10−5 eV/atom,最大作用力收敛标准为0.05 eV/Å,最大应力收敛标准为0.1 Gpa,最大位移收敛标准0.002 Å。
为了使模型计算结果更加准确,我们测算了不同U值作用下的LaFeO3带隙值。结果表明,当U = 5.25 eV时,LaFeO3的带隙为2.037 eV,与实验值2.1 eV [24] 最接近。选择合理的U值有利于体系的计算结果更符合实际,因此在所有的计算中都设定U = 5.25 eV。LaFeO3优化后,创建以Fe-O为端面的LaFeO3 (010)晶面并添加10 Å的真空层模拟周期性边界条件。Slab结构采用1 × 2 × 1超胞模型,包含两个Fe-O层和两个La-O层。结构如图1所示。吸附能的计算表达式为
。其中,
表示吸附体系的总能量,
表示LaFeO3 (010)表面的能量,
表示NO2分子的能量。式中,吸附能为负值时,表示吸附可能发生。吸附能的大小是评价吸附稳定性的重要标准。
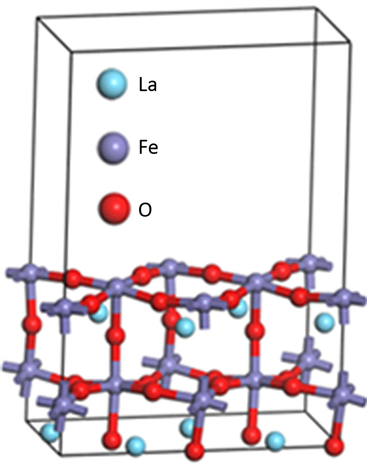
Figure 1. The side view of the FeO-terminated LaFeO3 (010) surface
图1. 以FeO为终端的LaFeO3 (010)表面
3. 结果和讨论
3.1. NO2在Fe-O端面LaFeO3 (010)表面的吸附
首先,我们计算了单个NO2分子的基本性质。构建尺寸为10 × 10 × 10 Å3的立方晶胞对NO2分子完成几何优化及性质计算,优化后的NO2分子的O-N-O键角为133.291˚,N-O键长为1.23 Å,这与实验值134˚和1.20 Å基本一致 [25]。为了获得稳定的LaFeO3 (010)表面结构,对构建的Slab模型进行了结构优化。在驰豫后的LaFeO3 (010)表面,探究NO2在以Fe-O为端面的LaFeO3 (010)表面上的4种吸附构型,分别为N原子在表面Fe原子上的顶位吸附(M1)、N原子在表面O原子上的顶位吸附(M2)、O原子在表面Fe原子上的顶位吸附(M3)以及N原子在表面的桥位吸附(M4)。初始吸附构型及优化后的结构和计算参数如图2及表1所示。4种吸附构型中,吸附能大小顺序为M3 (0.590 eV) < M2 (0.654 eV) < M4 (0.853 eV) < M1 (0.905 eV),构型M4和M1的吸附能属于化学吸附的能量范围,这表明构型M4和M1发生了稳定的化学吸附。相较于构型M4,M1的吸附能数值更大,吸附反应更加剧烈。另外,吸附后的NO2分子和LaFeO3 (010)表面之间的作用距离为M2 < M3 < M4 < M1。在构型M1和M4中,相互作用的N原子和Fe原子之间的距离D分别为2.208 Å和2.268 Å,这说明N原子和Fe原子之间形成了化学键。同时,吸附后构型M1、M3和M4中NO2分子的O-N-O键角发生了明显的改变。以上结果表明,构型M4,M1发生了稳定的化学吸附,其中构型M1为最稳定的吸附构型。

Table 1. The calculated data of 4 configurations of NO2 adsorption on LaFeO3 (010) surface
表1. NO2在LaFeO3 (010)表面吸附4种构型的计算参数
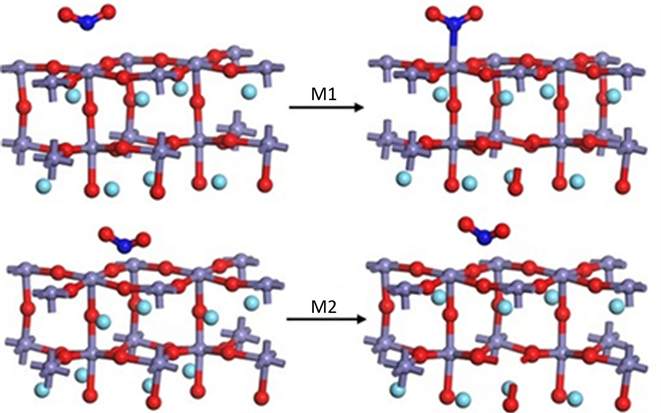

Figure 2. The adsorption configurations of NO2 on the LaFeO3 (010) surface
图2. NO2在LaFeO3 (010)表面的吸附构型
3.2. Millikan电荷分析
为了量化吸附过程中NO2和LaFeO3 (010)表面之间的电荷转移,分析了NO2分子在吸附前后的Millikan电荷布居,见表2。Millikan电荷分析中负号表示电子从吸附剂转移至气体分子,而正号表明电子从气体分子转移到吸附剂 [26]。计算结果表明,所有吸附构型中NO2分子的Millikan电荷均为负数,这意味着所有吸附构型在吸附过程中电子都是从LaFeO3 (010)表面转移到NO2分子,即吸附反应中NO2分子充当电子接受体。
从表2中可知,在4种吸附构型中,M1中NO2分子的净电荷在吸附反应后从0变化为−0.29 e,净电荷转移量最大,表明吸附作用最强烈,这与吸附能表现的结果相一致。其中N原子的电荷从0.42 e大幅减小到0.21 e,O原子的电荷均从−0.21 e变化为−0.25 e。这表明在吸附反应过程中,电子首先从LaFeO3 (010)表面转移到NO2分子中的N原子,接着部分电子进一步转移至NO2中的O原子。NO2中电子从N原子进一步转移到O原子主要归因于O原子的电负性比N原子的强,这与吸附前NO2分子中N原子显示正电荷而O原子显示负电荷一致。对于构型M4,NO2分子总的Millikan电荷为−0.28 e,略小于M1中的电荷转移量,这表明NO2和LaFeO3 (010)表面之间的作用较M1弱。构型M2和M3的净电荷转移量分别为−0.16 e和−0.23 e,这不足以在NO2和LaFeO3 (010)表面之间形成化学键。综上结果表明,构型M1和M4中NO2分子的Millikan电荷大,NO2和LaFeO3 (010)表面之间发生了强的化学吸附,并且M1为最稳定的吸附构型,这与吸附能的分析结果保持一致。
3.3. 电子定域和态密度分析
电子定域函数(ELF)可以清晰和定量描述化学键的强弱,因而在固体体系的研究中得到了广泛的应用。本文中为了更加直观地说明吸附反应中的成键情况,计算了最稳定吸附构型M1的电子定域函数,如图3。从图3中可以观察到NO2分子中的N原子和LaFeO3 (010)表面的Fe原子之间存在很强的电子定域性,
Table 2. Millikan population analysis for NO2 of NO2/LaFeO3 (010) adsorption system
表2. NO2/LaFeO3 (010)吸附体系中NO2的密立根布居分析
这说明N原子和Fe原子之间形成了强的化学键。此外,通过比较LaFeO3 (010)表面吸附有NO2分子的Fe原子和未吸附NO2分子的Fe原子,我们发现Fe原子表面吸附了NO2分子后自身的电子定域性发生了很大的改变,这说明NO2分子的吸附极大地影响了Fe原子的电子结构,吸附反应中NO2分子和LaFeO3 (010)表面之间发生了强烈的相互作用并且在Fe原子和N原子之间形成了强的化学键。

Figure 3. The electron localization function of M1 configuration
图3. M1构型的电子定域函数
NO2分子在LaFeO3 (010)表面上的吸附机理是本文研究的重点之一,因此我们计算了最稳定吸附构型M1的相关态密度,包括吸附反应前后NO2分子的态密度(图4)、吸附前后LaFeO3 (010)表面与NO2分子直接作用的Fe原子的态密度(图5)。从图4中我们发现吸附反应发生后,NO2分子的态密度发生了很大的变化,主要表现在态密度的峰位置向低能量区域发生移动,这表明在吸附过程中NO2的能量变得更低,NO2分子的结构更加稳定。进一步比较图4(a)和图4(b)可知,吸附后NO2分子在费米能级附近的态密度发生了明显的改变,峰位态密度值减小,而对应的能量范围相应变宽,这说明吸附反应后,NO2分子中价电子的离域性更强,NO2分子参与了成键。图5为吸附前后LaFeO3 (010)表面Fe原子的态密度图。从图5(a)可以看出,Fe原子的态密度在−7.06 eV、2.47 eV处出现了明显的尖峰,表明Fe原子本身的核外电子具有非常强的局域性。比较图5(a)和图5(b)我们发现,吸附后LaFeO3 (010)表面Fe原子的态密度在−8 eV和8 eV能量范围内的峰变化不大,但向低能量方向发生了轻微移动,而在−11 eV至−9 eV的能量范围内新分裂出一个峰,最新出现的峰与吸附后NO2分子的态密度峰位(图4b)能量范围重叠,这是由于表面Fe原子和N原子之间形成了Fe-N键的缘故。
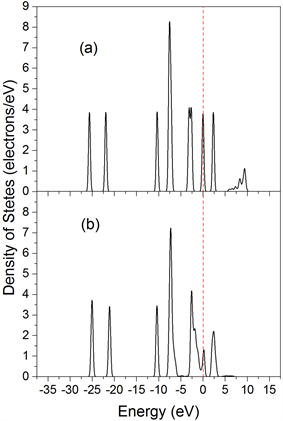
Figure 4. The DOS of the free and adsorbed NO2 with preferable M1 configuration: (a) before adsorption; (b) after adsorption
图4. M1构型中NO2分子吸附前后的态密度:(a) 吸附前,(b) 吸附后
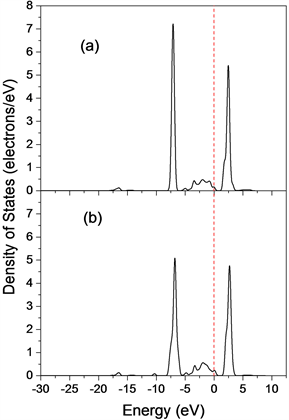
Figure 5. The DOS of Fe site before and after NO2 adsorption: (a) before adsorption; (b) after adsorption
图5. 表面Fe原子吸附前后的态密度:(a) 吸附前,(b) 吸附后
为了进一步探究NO2分子中N原子和LaFeO3 (010)表面Fe原子之间在吸附反应中的作用机理,分析吸附原子的分波态密度是必要的。首先,通过对比图6中N原子2p轨道的态密度和吸附后NO2分子的态密度(图4(b))分布,我们发现NO2分子中能量为−6.75 eV处的尖峰主要成分为N原子的2p轨道,这说明N原子中主要参与形成化学键的是2p轨道。另外,从图6中明显地观察到吸附后的N原子的2p轨道和表面Fe原子的3d轨道大幅重叠,并且局域尖峰的峰值减小,这表明吸附反应使得N的2p轨道和Fe的3d发生了强烈杂化,即N原子和表面Fe的原子发生了强化学作用,形成了新的化学键。在NO2分子中N原子的2p轨道和LaFeO3 (010)表面Fe原子的3d轨道之间观察到4个主共振峰,峰位能量分别为−10.37 eV、−7.15 eV、0.86 eV、2.34 eV。这些共振峰显示NO2分子中N原子和LaFeO3 (010)表面Fe的原子之间形成了Fe-N键,这与上述分析分析结果保持一致。综上分析可知,构型M1中NO2分子和LaFeO3 (010)表面之间的吸附机理为N原子2p轨道和Fe原子3d轨道之间的强烈杂化形成了Fe-N化学键。
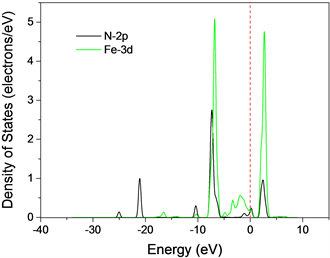
Figure 6. The PDOS of Fe 3d and NO2 2p orbital after the NO2 adsorption
图6. NO2 2p原子和表面Fe 3d原子吸附后的分波态密度

Figure 7. The X2+ (X = Ni, Cu, Zn) substitution position
图7. X2+ (X = Ni, Cu, Zn)取代位置示意图
3.4. X2+ (X = Ni, Cu, Zn)掺杂对LaFeO3 (010)表面吸附NO2的影响
基于之前的报道,本节我们研究了X2+ (X = Ni, Cu, Zn)掺杂对LaFeO3 (010)表面吸附NO2性能的影响。掺杂结构为分别用一个X2+取代LaFeO3中的一个Fe3+,取代位置如图7所示。
考虑到体系包含原子数目多、计算耗时长,因此我们仅通过计算X2+掺杂后M1构型中的相关参数探究X2+掺杂对LaFeO3吸附NO2性能的影响。离子掺杂后构型M1的相关计算结果见表3。
Table 3. NO2 adsorption on X2+ (X = Ni, Cu, Zn) doped LaFeO3 (010)
表3. X2+ (X = Ni, Cu, Zn)掺杂的LaFeO3 (010)表面吸附NO2的计算结果
从表中可知,向纯净的LaFeO3 (010)表面分别掺杂Ni2+、Cu2+和Zn2+离子后体系的吸附能从−0.905 eV分别变为−0.926 eV、−0.915 eV、−1.185 eV。吸附能越负,表明吸附作用越强。因此,掺杂Ni2+、Cu2+和Zn2+均能有效增强对NO2分子的吸附作用,其中Zn2+掺杂对LaFeO3 (010)表面吸附NO2的影响最大。此外,掺杂体系吸附反应后的Fe-N键长分别为2.179 Å、2.178 Å和2.164 Å,这比未掺杂体系中的键长更短,即吸附后形成的化学键更强。同时NO2分子的Millikan电荷变得更负,表明NO2分子和LaFeO3 (010)表面之间的

Figure 8. The PDOS of doped ions: (a) Fe3+; (b) Ni2+; (c) Cu2+; (d) Zn2+
图8. 取代位置离子的分波态密度:(a) Fe3+;(b) Ni2+;(c) Cu2+;(d) Zn2+
作用更强,转移的电子数目更多。通过分析不同掺杂离子的态密度,能够进一步解释离子掺杂对吸附作用的影响,图8显示了吸附反应后掺杂位不同离子的分波态密度。从图8中可以看到Fe3+、Ni2+、Cu2+、Zn2+的态密度存在明显的差异,尤其是3d轨道态密度,Ni2+、Cu2+和Zn2+的3d电子能量更低,表明吸附后结构更加稳定。综合上述结果可知,X2+掺杂能够增强Fe原子和N原子之间的作用,即X2+ (X = Ni, Cu, Zn)掺杂能增强LaFeO3 (010)表面对NO2的吸附性能。这主要归因于Ni2+、Cu2+、Zn2+与Fe3+在3d轨道电子数、电负性和离子半径等方面的差异。
4. 结论
本文基于密度泛函理论方法研究了正交相LaFeO3 (010)表面和NO2分子之间的吸附作用及X2+ (X = Ni, Cu, Zn)掺杂和氧空位缺陷对吸附性能的影响。计算结果表明NO2分子在正交相LaFeO3 (010)表面吸附的最佳吸附发生在NO2分子中的N原子和LaFeO3 (010)表面的Fe原子之间,吸附反应后形成了新的Fe-N化学键。反应中电子从LaFeO3 (010)表面转移至NO2分子,NO2分子为电子的受体。态密度的计算结果表明吸附反应机理为Fe原子的3d轨道和N原子的2p之间存在强烈杂化。X2+ (X = Ni, Cu, Zn)掺杂后,LaFeO3 (010)表面对NO2分子的吸附作用变得更强,吸附能更大。Zn2+掺杂对吸附反应的作用更加明显。