1. 引言
稀土配合物的发光致光现象早在二十世纪四五十年代就已被观察到,Weissman发现稀土配合物中因为含有某些具有共轭体系的有机配体,可用近紫外光激发而发射荧光 [1],在光致发光、激光材料及光导材料等方面 [2] 都具有很大的研究价值。此后该类配合物的研究就成为一个热点。
稀土离子发光具有谱带窄、荧光寿命长、发光强度高的优点 [3],其特殊的4f电子结构决定了发光特性。与有机配体配合后,可改变稀土离子在紫外区吸收系数小、发光不强的缺陷,含有共轭体系的有机配体可有效的吸收能量传递给发光中心稀土离子,使配合物具有良好的发光性能 [4]。铕配合物被认为是一种较好的红光材料 [5],Eu3+离子特征发射在红光区域,且发光强度好、色纯度高、化学稳定性好 [6],然而在发光材料领域,研究发现:由于三价稀土金属离子的f*-f跃迁属宇称禁阻,因而跃迁较弱且激发态易失活,导致荧光极弱 [7],提高荧光粉的发光亮度主要依靠调整第一配体的结构 [8],故选择合适的配体可以强化配合物的发光性能。师奇松等 [9] 人通过沉淀法将2-噻吩甲酰三氟丙酮作为第一配体制备出Eu(TTA)3(TPPO)2三元配合物;何玉鲜等 [10] 采用对羟基苯甲酸合成了具有的新型结构的稀土配合物。羧酸是除β-二酮之外的另一类良好的配体 [11]。β-二酮类配合物有相当高的发光强度,但在实际应用中却有许多受到限制的地方,当前稀土配合物面临加工性能较差 [12],制备工艺耗时较长等问题,所以为了获得性能更好、收率更高的铕配合物,研究不同的第一配体、制备方法及条件具有重大意义。本实验以三苯基氧膦为协同配体,选用不同的β-二酮类化合物或芳香羧酸类化合物作为第一配体,对Eu3+配合物进行合成工艺优化。采用紫外光谱分析、红外光谱分析、分子荧光光谱等现代仪器,对合成稀土铕配合物进行含量测定、结构和发光性能鉴定。
2. 材料与设备
2.1. 材料与试剂
材料:六水合氯化铕(分析纯):萨恩化学技术(上海)有限公司;三苯基氧膦(分析纯):萨恩化学技术(上海)有限公司;2-噻吩甲酰三氟丙酮(分析纯):上海麦克林有限公司;乙酰丙酮(分析纯):永华化学科技(江苏)有限公司;二苯甲酰甲烷(分析纯):上海麦克林有限公司;对溴苯甲酸(分析纯):上海麦克林有限公司;苯甲酸(分析纯):上海来泽精细化学品研究所出品;水杨酸(分析纯):广东东莞市聚鹏化学有限公司;溴化钾(优级纯):上海凌峰化学试剂有限公司。
试剂:氢氧化钠(分析纯):杭州萧山化学试剂厂;三氯甲烷(分析纯):广东西陇化工股份有限公司;无水乙醇(分析纯):杭州青辰化学试剂厂;使用水均为蒸馏水。
2.2. 仪器与设备
数显恒温磁力搅拌器07-HWS-2:杭州仪表电机有限公司;EXCEL微波化学反应系统:上海屹尧仪器科技发展有限公司;傅立叶红外光谱仪BRUKER TENSOR-27 FT-IR型:德国Bruker公司;紫外分光光度计UV-2450:日本岛津公司;分子荧光光谱仪F-4600型:日本日立公司;三用紫外线分析仪ZF-6型:上海嘉鹏科技有限公司;电热恒温鼓风干燥箱DGG-9140A:上海森信实验仪器有限公司;旋转蒸发器RE-3000:上海亚荣生化仪器厂;超声波清洗机JK-100:合肥金尼克机械制造有限公司。
3. 实验方法
3.1. 稀土铕配合物的合成
以三价稀土铕离子(Eu3+)为中心配位离子,β-二酮类化合物或芳香羧酸类化合物为第一配体,三苯基氧膦为第二配体,合成三元稀土铕配合物。本实验采用水浴法分别用2-噻吩甲酰三氟丙酮(TTA)、二苯甲酰甲烷(DBM)、水杨酸(SA)、乙酰丙酮(Hacac)、对溴苯甲酸(PBrBA)、苯甲酸(BA)六种化合物作为不同的第一配体进行合成实验。
3.1.1. 制备含TTA的铕配合物
根据氯化铕:2-噻吩甲酰三氟丙酮:三苯基氧膦 = 1:3:2的摩尔比,将0.3331 g 2-噻吩甲酰三氟丙酮和0.2782 g三苯基氧膦溶于8 mL无水乙醇。称取0.1833 g EuCl3∙6H2O溶于2 mL无水乙醇,在磁力搅拌的条件下,滴加入上述混合液。滴加2.0 mol/L的氢氧化钠溶液(调节pH至6~7),65℃水浴搅拌加热4 h后,静置10 h,常压过滤,用乙醇和蒸馏水分别洗涤两次 [3]。最后放入旋转蒸发仪,60℃抽真空干燥30分钟,至恒重。
3.1.2. 制备含DBM的铕配合物
按照氯化铕:二苯甲酰甲烷:三苯基氧膦 = 1:3:1的摩尔比 [13] 计算,准确称取0.3365 g二苯甲酰甲烷和0.1627 g三苯基氧膦,0.1832 g EuCl3∙6H2O,其他操作同3.1.1。
3.1.3. 制备含SA的铕配合物
按照氯化铕:水杨酸:三苯基氧膦 = 1:3:2的摩尔比 [3] 计算,称取0.2071 g水杨酸和0.2781 g三苯基氧膦,0.1835 g EuCl3∙6H2O,其他操作同3.1.1。
3.1.4. 制备含Hacac的铕配合物
按照氯化铕:乙酰丙酮:三苯基氧膦 = 1:3:1的摩尔比 [14] 计算,称取0.1627 g三苯基氧膦溶于8 mL无水乙醇,量取乙酰丙酮0.154 mL,搅拌均匀,称取0.1832 g EuCl3∙6H2O溶于2 mL无水乙醇,其他操作同3.1.1。
3.1.5. 制备含PBrBA的铕配合物
按照氯化铕:对溴苯甲酸:三苯基氧膦 = 1:3:2的摩尔比 [15] 计算,准确称取0.3014 g对溴苯甲酸和0.2785 g三苯基氧膦,0.1833 g EuCl3∙6H2O,其他操作同3.1.1。
3.1.6. 制备含BA的铕配合物
按照氯化铕:苯甲酸:三苯基氧膦 = 1:3:2的摩尔比 [14] 计算,称取0.3366 g苯甲酸和0.2783 g三苯基氧膦,0.1863 g EuCl3∙6H2O,其他操作同3.1.1。
4. 方法优化
通过实验了解水热法耗时长,消耗功率大,且收率不算高。通过查阅文献 [16] [17],发现水热法可以用超声法和微波法等进行辅助,为提高制备效率及降低成本,以反应现象较明显的Eu(TTA)3(TPPO)2为目标铕配合物对实验方案进行优化。
4.1. 反应方法对收率的影响
按照氯化铕:2-噻吩甲酰三氟丙酮:三苯基氧膦 = 1:3:2的摩尔比计算,称取三份样品,分别采用水浴法、微波辅助法和超声法进行反应。
4.1.1. 水浴法
将三份样品溶于10 mL无水乙醇,具体参考3.1.1。
4.1.2. 超声辅助法
将混合液倒入圆底烧瓶,放置于超声反应器中,搅拌1 h并65℃超声水浴,后续操作同3.1.1。
4.1.3. 微波辅助法
将混合液倒入三口圆底烧瓶并放入磁石,用玻璃塞将左右瓶口封住,放入微波系统中,通循环水,打开磁力搅拌,将程序设定为四个步骤,每个步骤为功率200 W、反应5 min,并打开磁力系统。反应停止,待系统冷却至室温,取下圆底烧瓶,冷却。后续操作同3.1.1。
4.2. 单因素对收率的影响
通过4.1实验得到效率较高的合成方法,对该方法进行功率、温度、反应时间、料液比等的单因素实验。最终得到合成Eu(TTA)3(TPPO)2的最佳条件。
5. 表征方法
5.1. 紫外–可见吸收光谱
使用UV-2450紫外分光光度计对六种配体的吸收峰进行观察,判断配合物合成情况。
5.2. 红外光谱
采用 BRUKER TENSOR-27 FT-IR型傅里叶红外光谱仪对六种配合物的官能团和结构进行鉴定。在4000~400 cm−1范围内,用KBr压片法测定配体与配合物的红外光谱。需特别注意水分的去除,水对测红外有干扰。
6. 发光性能测定方法
采用日立F-4600型分子荧光光谱仪,扫描所制备的六种配合物在氯仿中(1 × 10−4 mol/L) 200~800 nm范围的荧光光谱,对配合物的荧光强度进行测试。
7. 铕的含量测定方法
采用铂金埃尔默Elan DRC-e型电感耦合等离子体体质谱仪,测定样品中稀土金属铕的含量。
8. 结果与分析
8.1. 实验结果
8.1.1. 收率计算
将上述恒重后的产物冷却至室温,称重,将样品所测得的质量除以根据摩尔比1:1计算出的EuCl3∙6H2O的理论产量:
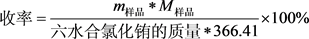 (1)
(1)
由表1可知,第一配体为2-噻吩甲酰三氟丙酮(TTA)时,铕配合物收率最高。

Table 1. Preparation of europium complex
表1. 不同第一配体制备铕配合物的结果
8.1.2. 紫外–可见吸收光谱
由不同配体制备的六种配合物在氯仿中(1 × 10−4 mol/L)的紫外–可见吸收光谱如图1。
图1(a)与文献 [18] 所示光谱图相同。配合物较易溶于氯仿等挥发性溶剂,在200~600 nm范围内有三个较大吸收峰,HTTA有个269 nm的吸收峰。配体及配合物在紫外光区均有较强的吸收,来源于π~π*及n~π*跃迁。在配合物的紫外吸收光谱上,配体TPPO的峰位形成配合物后消失,配体在290 nm附近峰位移至343 nm,大大增强了配合物的吸收强度,表明形成比配体更大的共轭体系。配合物峰形与第一配体相似,说明紫外吸收主要来自第一配体的贡献。峰位及峰强度的改变证明配体与稀土离子形成了键合作用。
图1(b)、图1(c)、图1(d)存在与文献 [19] 所示光谱图相似的波峰,分别显示二苯甲酰甲烷、水杨酸、乙酰丙酮为第一配体制成配合物的紫外吸收光谱。峰位分别为348、313、344 nm,都处于本试验所需波段,但相比于图1(a)峰高过低。
图1(e)、图1(f)分别显示对溴苯甲酸和苯甲酸为第一配体制成配合物的紫外吸收光谱。都在238 nm有较大吸收峰,图1(e)在344 nm有一个较小波峰。
8.1.3. 红外吸收光谱
配合物的红外光谱特征吸收峰和配体的特征吸收峰明显不同。
如图2,配合物的红外吸收光谱图显示了与文献 [18] 一样的波峰。自由配体HTTA有2个羰基不对称伸缩振动峰,靠近噻吩环的C=O为1656 cm−1附近,靠近电负性强的三氟甲基的C=O为1620 cm−1附近。
羰基不对称伸缩振动峰在形成配合物后分别红移至l611 cm−1、1632 cm−1、1629 cm−1、1637 cm−1、1590
cm−1、1707 cm−1,羰基的频率发生低频位移,出现红移现象,说明稀土金属和羰基配位。配位后的C=C双键位于1535 cm−1、1548 cm−1、1485 cm−1、1439 cm−1、1541 cm−1、1548 cm−1,与配位前相比后移。配体TPPO的P=O伸缩振动峰由1190 cm−1附近分别红移至1173 cm−1、1161 cm−1、1157 cm−1、1188 cm−1、1169 cm−1、1166 cm−1附近,同时六种配合物在540 cm−1附近都出现了Eu-O的伸缩振动吸收峰,进一步证实氧原子与铕离子配位。配合物在3450 cm−1附近有一个羟基反对称伸吸收,-OH的峰值远小于六水合氯化铕,这些说明稀土离子与配体HTTA、TPPO发生了配位。
8.1.4. 荧光光谱分析
首先用紫外最大吸收波长作为激发光,快速扫描发射谱,然后选择发射谱中最强信号对应的波长作为检测波长,扫描激发谱 [8]。
由不同配体制备的六种配合物在氯仿中(1 × 10−4 mol/L) 200~800 nm范围的荧光光谱如下。
由图3荧光吸收光谱可知,羧酸配体与铕配合物的荧光光谱均以配体的强宽峰为主,其610至620 nm的发射峰,属于Eu3+的5D0→7F2电偶极跃迁,发出红色荧光,同时也是Eu(III)离子的特征发射峰,其强度受配位环境的影响,在五个Eu3+的荧光发射峰中,以5D0→7F2跃迁的相对荧光强度最强 [20],表明Eu(III)中心离子没有反演对称中心。
其余配合物都只有在400nm左右的一条窄吸收峰,只有Eu(TTA)3(TPPO)2的吸收峰在该范围内为616.2 nm,且最大波峰的形状与文献 [3] 的谱图相同。由配合物的发射光谱可以看出616 nm发射峰又窄又强,表明配体最低三重态能级与Eu3+的发射能级5D0匹配较好,不存在反演中心,色纯度较高。
由荧光发射光谱可知,200~300 nm的范围内只有Eu(TTA)3(TPPO)2存在较宽强吸收峰。其余均低于400 nm左右的峰值。
8.1.5. 结果
结合上述测试分析结果,水浴法制备的六种配合物中,以2-噻吩甲酰三氟丙酮为第一配体制备的三元配合物Eu(TTA)3(TPPO)2,发光性能最好,荧光强度最大。
8.2. Eu(TTA)3(TPPO)2的ICP-MS分析
称取0.1141 g样品检测,以标液浓度为0、1、5、10、15、20 ppb。
得到线性方程y = 135.649X + 1517.61 (R2 = 0.999584),测得Eu含量为115.57 g/kg。
分析结果见表2。
Table 2. Element concentration in Eu(TTA)3(TPPO)2
表2. Eu(TTA)3(TPPO)2中的元素浓度
8.3. 方法优化结果
8.3.1. 反应方法对收率的影响
根据公式(1)计算收率,结果如表3。
Table 3. Effect of reaction method on yield
表3. 反应方法对收率的影响
由上述看,虽然微波法在收率上较低,但与超声法所得收率相差不大,且从反应时间上看,微波辅助法的用时仅为20分钟,远低于其余两种。所以本实验铕配合物的最佳制备方式是微波辅助法。
8.3.2. 单因素对收率的影响
由上述可知,在水浴法,超声法,微波法之中,微波法制备配合物Eu(TTA)3(TPPO)2,但是产率只有65.31%。为了制备出产率更高的LED发光材料,对微波功率、反应时间、料液比进行单因素实验。因该实验室微波系统的温控系统受损,且温度主要受微波功率的影响,故本次实验不考虑温度因素对收率的影响。
(1) 功率
设定微波反应时间为20 min,料液比为1:25。称取0.0366 g EuCl3∙6H2O、0.0667 g 2-噻吩甲酰三氟丙酮、0.0557 g三苯基氧膦溶于5 mL无水乙醇,在磁力搅拌的条件下,滴加2.0 mol/L的氢氧化钠溶液,调节pH至6~7,分别在100 W、150 W、200 W、250 W下进行反应。反应结束后,进行收率计算,如下表4:
Table 4. Effect of microwave power on the yield ofEu(TTA)3(TPPO)2
表4. 微波功率对Eu(TTA)3(TPPO)2收率的影响
在微波功率为200 W时,配合物收率达到最大,为65.31%,故在200 W左右最为合适。
(2) 时间
设定微波反应功率为200 W,料液比为1:25。制备相同的混合液同6.2.1,分别在10 min、15 min、20 min、25 min、30 min下进行反应。结果如下:
Table 5. Effect of microwave time on the yield of Eu(TTA)3(TPPO)2
表5. 微波时间对Eu(TTA)3(TPPO)2收率的影响
由上述表5数据可得,收率随着时间增加而赠加,在20 min的时候达到最大值65.31%,为最佳反应时间条件。随着时间继续加长,产率开始有所下降。反应时间的加长,能让反应更完全,但保持的时间的加长,会使配合物发生副反应,造成产率下降。
(3) 料液比
设定微波反应功率为200 W,时间为20 min。称取同6.2.1的等量样品,分别溶于1:15、1:20、1:25、1:30、1:50无水乙醇,调pH,在20 min条件下进行反应。结果如下:
Table 6. Effect of feed-to-liquid ratio on the yield of Eu(TTA)3(TPPO)2
表6. 料液比对Eu(TTA)3(TPPO)2收率的影响
如表6所示,在料液比为1:15时,表现为产率最高,为68.63%,之后随着溶剂用量的增加,收率下降,可能因为过量乙醇导致结晶无法析出。当料液比小于1:15时,溶液无法浸没样品,故不考虑更小的料液比。
8.3.3. 结果
经过优化实验得出,采用微波法辅助反应制备配合物Eu(TTA)3(TPPO)2的最佳工艺条件为:微波反应时间20 min,功率200 W,料液比1:15。在此反应条件下Eu(TTA)3(TPPO)2的收率可达68.63%,此制备方法相比传统的制备方法,具有耗时少、能耗低和提取效率高等优势,在稀土有机配合物发光材料的制备应用中,具有一定的优势。
9. 结论
(1) 通过水浴法制备出的稀土发光材料中,Eu(TTA)3(TPPO)2产量高、荧光性能好,结果表明Eu(TTA)3(TPPO)2配合物符合当前研究所需。
(2) 运用微波法、超声法、水浴法辅助制备Eu(TTA)3(TPPO)2发光材料。较水浴法来说,微波法具有耗时短、耗能低、产率高等优点,是制备Eu(TTA)3(TPPO)2发光材料的新型方法。
(3) 首次对微波法进行了反应物料液比、微波功率、反应时间三因素的实验优化,得出最优工艺:微波反应时间20 min,功率200 W,料液比1:15。产率为68.63%。较优化之前75.35%的产率,只降低了7%,用时却缩短为原来的十二分之一。
基金项目
浙江省高校实验室工作研究项目(YB201845)。
NOTES
*通讯作者。