1. 引言
近年来,第一性原理高通量计算(ab-initio high-throughput computational methods)在预测新材料和优化材料的属性等方面被证明是一种强大而且成功的方法。例如:多组分晶体、合金相图的成功预测 [1] [2],太阳能电池材料的电极透明度和电导率的优化,得到更好的电流–电压特性 [3]。然而,大量的计算资源需求成为了高通量计算的瓶颈。当化合物可能的结构搜索空间变得很大,元胞的结构变得很复杂时,即使采用高效的Kohn-Sham密度泛函理论(KS-DFT),所需的计算量对于有限的计算资源来说也是一项挑战。人们希望有一种更快的方式来预测新材料的物理属性,而无需求解KS-DFT方程。最近,机器学习(ML)技术逐渐被人们重视。对于预测新材料的物理属性研究方面,机器学习技术和传统的基于第一性原理计算的本质区别在于,机器学习摒弃了传统计算方法中通过求解多电子体系薛定谔方程来预测材料的物理属性,而是直接从大量已知的物理数据出发,通过多层次神经网络的运算,找到不同材料的不同物理属性的内在网络关系,从而迅速地得到未知新材料的物理属性。基于机器学习的计算效率很高,模型训练结束后,预测给定材料的特定属性通常仅需要几秒。此外,机器学习在预测分子特性,过渡态,表面反应等领域也有了较大发展,出现了一系列精准的模型 [4] [5] [6] [7]。
深度神经网络(DNN)模型可以直接从输入表示中学习,例如文本的数字编码,图像的颜色像素等,无需研究人员考虑如何进一步描述数据 [8],从而省略了建立传统机器学习模型中所需的特征工程等人工步骤。由于这种优势,深度学习近些年在计算机科学领域获得了极大的关注,在计算机视觉 [9] 、语音识别 [10] 和文本处理领域 [11] - [18] 取得了突破性进展。尽管深度学习模型在上述应用中取得了巨大成功,但材料科学中深度学习的发展还处于早期阶段。本文采用深度学习快速预测晶体性质,利用由多个神经层组成的DNN模型学习数据间的关系,对深度学习模型在材料结生成焓的预测做了深入的研究和讨论。
2. 神经网络预测晶体生成焓
我们利用深度学习,直接从元素的组成中学习材料的属性,消除了当前需要手动特征工程的局限性。它仅将元素组成作为输入,并利用网络自动捕获基本化学关系来预测生成焓。
人工神经网络由许多人工神经元连接而成。它们接受输入,然后根据输入调整自身的激活状态,并根据输入和激活状态产生输出。通常,神经元j除了从它的上级神经元接收到的输入
以外,它的状态还包含:自身的激活状态
、阈值 、激活函数f和输出函数
。
、激活函数f和输出函数
。
激活函数f用于计算时刻t到t + 1时的激活状态,取决于
和净输入 的关系:
的关系:
(1)
神经元之间相互连接,每个连接将神经元i的输出传递给神经元j的输入。因此,i是j的上级,j是i的下级,连接会被分配一个权重
。因此,上级神经元的输出
计算输入到神经元j的输入
取决于其所有上级(有连接的)神经元的输出和对应连接的
:
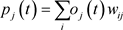 (2)
(2)
当需要添加一个偏差项时,方程变为:
 (3)
(3)
其中
为偏差。
训练一个神经网络本质上是在所有的可能的模型参数中(超空间中)选择能正确映射输入与期望的模型。有许多算法可以训练神经网络,其中绝大多数可以被视为是一种基于统计估计的优化理论。
绝大多数是采用反向传播来计算梯度下降。只需计算损失函数的梯度然后根据该梯度调节网络的参数,使结果更靠近最优处。反向传播的主要特征是迭代,循环更新网络参数,直到网络能够有效完成正在训练的任务,将输入准确的映射到期望的输出。反向传播的权重更新可以通过随机梯度下降使用以下等式完成:
 (4)
(4)
其中
是学习率,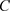 是损失函数,
是损失函数, 是一个随机数。损失函数的选择取决于学习类型和激活函数。
是一个随机数。损失函数的选择取决于学习类型和激活函数。
机器学习中的学习率
是一个超参数,它决定神经元新获取的信息在多大程度上覆盖旧信息。
的选择很重要,因为较高的值会导致过强的变化,导致错过最小值,而过低的速度会不必要地减慢训练速度。
应始终小于1,否则网络将不会收敛,通常
。 的最佳值因模型和数据而异,所以为了加速误差最小化,提高可靠性,
通常在训练期间根据学习速率表或通过使用自适应学习速率而变化。目前,我们有很多优化函数可使用,比如Adagrad,Adadelta,RMSprop,Adam等,它们通常内置于深度学习库中。其中RMSprop算法基于Rprop,是一个专为神经网络设计的优化算法,近年来在自适应学习方法中受到广泛关注。
最优问题的梯度的幅度可能变化很大,这时我们找到模型合适的学习率就变得困难。如果使用全批量学习模式,只能计算模型的平均梯度来确定学习率。这种方式对于高梯度的鞍点或极值点位置没什么问题,因为只要我们有足够的迭代次数,即使每一步的步进距离很小,我们最终还是能到达最优点。然而,如果模型的最优点附近梯度很小,很可能被越过。Rprop试图让学习率根据模型自动变化,在梯度小的区域变小,在梯度大的区域变大。该方法结合了梯度和权重模型,每次迭代都会根据特定的权重自动调整步长。首先,查看最后两步的梯度,如果它们的符号相同,证明搜索的方向正确,可以轻微的增加步长;如果符号相反,说明搜索的步长过大了,跳过了一个局部最小,因此要减小步长。最后为步长设置上下限,这个上下限需要根据模型的细节和数据集来设定。
Rprop的不足在于,当数据集非常大的时候,它不能执行小批量权重更新。例如:我们有10组数据,其中9个的梯度为0.1,1个为1。我们希望这个梯度能相互抵消,但是Rprop算法将连续增加9次权重,导致权重更大,步长变得不可控,因此本文采用RMSprop算法。
RMSprop的核心思想是保证权重是梯度的均方根。
 (5)
(5)
 (6)
(6)
其中
是平方梯度的平均,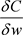 是损失函数权重的导数,h是学习率,
是个参数。
是损失函数权重的导数,h是学习率,
是个参数。
为了找到最佳的模型,我们先从两层网络开始,进行训练,测试精确度,之后逐步增加网络层数和神经元数,直到精确度不再显著增加。在两个拥有不同神经元的层之间需要加入dropout层 [19],防止过拟合的发生 [20]。在层数超过6层时,测试误差不再减小反而上升,因此我们认为此时的模型已经能学习到数据中必要的特征。进一步增加网络层数已经不能再提高精确度,但会显著增加模型训练的时间和过拟合的可能,所以我们的不再继续增加层数。我们还尝试了不同类型的激活函数,其中ReLU的表现最好 [20]。
本文模型所使用的275,778种化合物数据全部来自OQMD数据库,OQMD是一个广泛使用的高通量密度泛函(DFT)数据库,其中的数据包含了DFT计算过的晶体学参数,无机晶体结构数据库(ICSD)中生成焓的实验值等数据。我们选取每种成分的最低生成焓训练我们的预测模型,因为它们代表了最稳定的化合物,这使得我们的模型能够预测给定成分的基态结构的能量。275,778种化合物数据中,6.3%的材料由双元素构成,81.4%的材料由三元素构成,12.26%的材料由四元素构成。我们需要将化学式转化为模型能识别的表示方式:采用一个固定长度的向量,里面记录着每种元素原子数的比例值(当元素没有出现在化合物中时为0),我们的多层神经网络拓扑结构如图1所示。

Figure 1. Multilayer neural network topology. Elements are arranged according to the number of protons, and the normalized proportional value of elements is taken as the input vector of the input layer.
图1. 多层神经网络拓扑结构。将元素按质子数排列,将元素的归一化比例值作为输入层的输入向量
图2展示了实验过程中我们测试过的四种模型。标签32 × 2 + 16 × 2 + 8 × 1 + 1 × 1代表模型的架构为:两层32神经元网络加上两层16神经元网络加一层8神经元网络加一层1神经元网络(输出层)。由图可知当神经元层数增加,神经元数量增加,模型的误差下降。128 × 2 + 64 × 2 + 32 × 1 + 1 × 1结构的模型在我们所有的测试中误差是最低的。从图3我们可以看到,当我们继续增加神经元层,发现256 × 2 + 128 × 2 + 64 × 2 + 32 × 1 + 1 × 1结构出现了过拟合:即随着迭代次数的增加,训练集的误差(loss)在下降,但测试集的误差(val loss)不断上升。
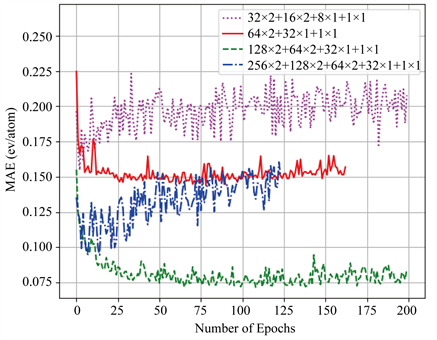
Figure 2. Relations between number of layers and error, the x-axis is the layer number model, and the y-axis is the error
图2. 层数与误差的关系,横坐标为层数模型,纵坐标为误差

Figure 3. Rraining situation of 256 × 2 + 128 × 2 + 64 × 2 + 32 × 1 + 1 × 1 structure.
图3. 256 × 2 + 128 × 2 + 64 × 2 + 32 × 1 + 1 × 1结构的训练情况
我们所使用的学习模型的最终结构见表1:
输入层为第0层,不同类型的全连接层和dropout的位置如表所示,dropout层用于防止过度拟合,它们不计为单独的层,模型中我们使用ReLU作为激活函数。
深度学习模型在许多应用中取得了巨大成功,但通常这些是训练数据相对丰富的应用,但数据量并非越多越好。一方面,复杂模型需要更多的训练数据,否则容易出现过拟合。另一方面,如果采用简单模型,即使给与大量的训练数据,也会因为模型无法描述复杂的关系而无法做出精准的映射,而且过量的数据会减慢训练速度。为了理解本深度学习模型的精度与数据集大小之间的关系,我们使用十折交叉验证的方法,比较了训练数据集大小对深度学习模型的准确性的影响。十折交叉验证是把样本数据随机分成10份,每次随机选择9份作为训练集,剩下的1份做测试集。训练结束后,重新随机选择9份再次进行训练,直至得到损失函数评估最优的模型和参数。本文中我们使用了四种大小的数据集分别进行研究:5000、10000、50000、275778。

Table 1. Structure of deep learning model
表1. 深度学习模型结构
由图4结果可知,该深度学习模型在数据集越大时,模型误差越小,精度越高。这说明我们的模型足够复杂,在大量的数据中能捕捉到其内在关系。我们将所有数据的90%作为训练集并经过多轮迭代后,模型逐渐收敛于某个最优解,见图5。
图6是使用25662个元素组成的测试集对模型进行10折交叉验证的结果。在其左图中,散点分布于y = x附近说明了深度学习模型的预测值和DFT计算值相接近。该模型预测的平均绝对值误差为0.075 eV/atom,表现出了很高的准确性。如右图所示,90%的预测误差小于0.180 eV/atom,80%的预测误差小于0.080 eV/atom。

Figure 4. Relationship between dataset size and accuracy of deep learning model
图4. 数据集大小与深度学习模型的准确性的关系

Figure 5. Performance of the model after several iterations. “loss” is the average absolute difference of the training set. “var loss” is the average absolute difference of the 10-fold cross-validation of the model with the test set consisting of 25,662 elements. The test set accuracy of the model converges to 0.075 eV/atom
图5. 模型在经过多次跌迭代后的表现。其中loss是训练集的平均绝对差值。var loss是使用25662个元素组成的测试集对模型进行10折交叉验证的平均绝对差值。模型的测试集精度收敛于0.075 eV/atom
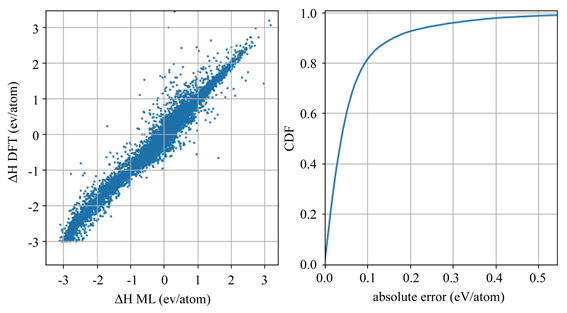
Figure 6. The results of a 10-fold cross-validation of the model using a test set of 25,662 elements. The x-axis of the left figure is the predictions of the neural network model, and the y-axis is the DFT calculation results. The right figure shows the cumulative distribution function (CDF)
图6. 使用25662个元素组成的测试集对模型进行10折交叉验证的结果。左图的x轴是神经网络模型的预测,y轴是DFT计算结果。右图为累积分布函数(CDF)
3. 结论
本文使用了机器学习算法在OQMD数据库的基础上建立了深度学习多层全连接网络,用来预测材料的生成焓。在优化后预测模型中,80%的预测误差小于0.080 eV/atom,平均误差为0.075 eV/atom,达到了DFT计算的精度,说明我们的模型可以用来预测未知材料化合物生成焓,之后我们将尝试将机器学习运用在其他材料属性的预测中。
致谢
感谢南京大学固体微结构物理国家重点实验室e-Science中心的计算支持。
基金项目
国家自然科学基金面上项目(批准号:11674054)、国家自然科学基金青年科学基金(批准号:11504182)、南京邮电大学校级科研基金资助项目(批准号:NY219087,NY220038)和南京大学固体微结构物理国家重点实验室开放项目资助的课题(批准号:M32025)。
NOTES
*通讯作者。