1. 引言
燃料电池是一种将化学能直接转化成电能的装置,具有清洁、高效等特点,但由于其从困难的制氢、储存、运输到复杂的电池设计、电极的低稳定性和阴极氧还原反应速率低下等原因依旧面临着巨大的挑战 [1]。 目前较为前沿的解决途径是采用具有相近能量密度、结构简单、阳极燃料可选择范围更广的金属空气燃料电池进行替代。但金属空气燃料电池仍属于燃料电池,阴极氧还原反应速率低下等问题同样成为了限制其发展的瓶颈,因此阴极催化剂的研究成为重点 [2] [3] [4]。 在阴极催化剂的研究当中,较为成熟的阴极催化剂均为铂、钯、银等贵金属,但这些贵金属价格昂贵、储量稀少,因此开发一种新型的高催化活性、低成本的ORR催化剂成为了当下研究的热点之一 [5]。
过渡金属氧化物氧化锰是一种十分常见的氧化物,其自身不仅拥有着较好的氧还原催化性能,还具备储量丰富、价格低廉、可选择性大等诸多优势,在很早之前就引起了科研工作者的研究兴趣。锰具有众多的氧化物形式,比如:MnO、MnO2、Mn3O2、Mn3O4、Mn5O8、MnOOH。通过研究比较,其催化活性排序为:Mn5O8 < Mn3O4 < Mn3O2 < MnO < MnOOH < MnO2。各类MnOx的性能差异主要是由于其电子结构和不同价态Mn的反应步骤所决定 [6]。 此外,锰氧化物的表面形貌与晶体结构对于其催化活性同样具有影响,其中最常见的是α-MnO2、β-MnO2和γ-MnO2。不同晶型的MnO2由于具有不同晶体结构和晶胞参数,因此其几何尺寸和形状也大不相同,进而导致其电化学性能也有很大差异。当下基本的观点认为MnO2的结构是以[MnO6]为基本单元,与相邻的连接形成单链或者双链结构,这些链结构的顶点与其它链共顶点形成隧道结构 [7] [8]。 α-MnO2是四方晶系,它的结构是双链连接的(1 × 1)、(2 × 2)的双隧道结构。β-MnO2同为四方晶系,它的结构则是单链连接的(1 × 1)隧道结构,这种结构相对来说空隙较小,不便于离子的扩散。γ-MnO2是斜方晶系,它的结构为(1 × 1)与(1 × 2)互相交错的双隧道结构。氧化锰不同晶型的催化活性顺序为γ-MnO2 < β-MnO2 < α-MnO2,这是由于其固有的隧道效应和尺寸效应来决定。Ogasawara [9] 小组研究了各种氧化物作为正极催化剂材料的性能,包括商用电解氧化锰、α-MnO2和β-MnO2的块体材料和纳米线材料、γ-MnO2、λ-MnO2、Mn2O3和Mn3O4。结果表明在测试的所有锰的氧化物中,α-MnO2具有最大的放电比容量。Haitao Zheng [10] 等也研究了氧化锰三种晶型对其电化学性能的影响,研究结果表明无论是300℃热处理前还是处理后,α-MnO2较之于β-MnO2、γ-MnO2具有最佳的电化学性质,极限电流密度达到7.03 mA/cm2。此外,不同晶型的氧化锰又具有着不同的纳米形态,例如纳米线、纳米棒、纳米球等。Oloniyo [11] 等的研究制备了α-MnO2纳米棒、α-MnO2纳米球和碳载α-MnO2纳米线,并对其催化活性进行了对比,结果证明了碳载α-MnO2纳米线性能效果最佳。
随着对锰氧化合物催化活性的深入研究,研究人员进行了各种尝试,其中包括对二元或复合锰氧化物掺杂一些金属元素便可制备出更加优异的催化剂。Jianpeng Li [12] 等人则研究了Fe、Co、Ni、V等不同离子掺杂氧化锰后其电催化性质的变化。结果表明,Ni、Co的掺杂使得氧化锰的电催化性能提升而Fe、V的掺杂导致氧化锰的电催化性质下降。孙明 [13] 等人将其它金属离子(Fe3+、Co2+、Ni2+、Cu2+等)引入到氧化锰的晶体之中,使得氧化锰内部的孔道内部结构发生了改变,进而制造了更多的表面缺陷,这些缺陷使得二氧化锰的二甲醚催化活性得到了很大的提升。陈素怡 [14] 等人将低价金属离子(Co2+、Ni2+、Cu2+)掺杂到二氧化锰之中,结果表明这些金属离子的掺杂不会使得氧化锰晶型、晶相和形貌发生改变并且均能够提高氧化锰的电催化性能,其提升的强度从大到小依次为Fe/α-MnO2 > Co/α-MnO2 > Ni/α-MnO2。
为了研究其它过渡金属掺杂氧化锰对其催化性能的影响,本文采用了水热法制备了同一掺杂比例下Co、Fe、Cu掺杂氧化锰纳米线的催化剂,并对其结构和性能做了表征及对比,获得了不同元素掺杂对氧化锰催化性能影响的结果。
2. 实验部分
为了确保所制备电催化剂的质量,本文使用的所有实验药品均为分析纯试剂且没有采用进一步的处理,配置溶液的所用水均为去离子水。其中20%的商用Pt/C催化剂是通过Johnson-Matthey公司所购买,其余的试剂均是通过Sigma-Aldrich所购买。
2.1. 催化剂的制备
本实验采用水热反应法,在制备纯α-MnO2的过程之中加入相应质量的硝酸盐来制备各种过渡金属掺杂的α-MnO2纳米线。首先,称量1.4725 g KMnO4溶于100 ml的去离子水中,后将其置于磁力搅拌器上不断搅拌。再称量2.2 g MnSO4·H2O和相应质量的硝酸盐分别溶于30 ml的去离子水中,可能并不能够完全溶解但至少保持其成为悬浮乳液状。将上述过程中后面的溶液使用胶头滴管逐滴的滴入到之前搅拌的溶液中。随后量取3 ml的浓硝酸滴入上述混合液中后保持搅拌1 h。将混合溶液放置反应釜中,密封后放入烘箱100℃反应24 h。水热反应后将样品抽滤,清洗至中性后真空60℃烘干6 h。最后,将烘干后的样品研磨并装入自封袋中贴好标签。本实验用于水热反应溶液中Mn/Co、Mn/Fe、Mn/Cu的原子比均为10:1,对应每种元素所需加入硝酸盐的质量分别为:0.374 g AgNO3、0.640 g Co(NO3)2·6H2O、0.532 g Fe(NO3)3、0.413 g Cu(NO3)2。
2.2. 电化学性能表征
本文所采用的电化学测量仪器是ATA-IB型旋转圆盘电极,工作电极采用的是直径1 mm的玻碳电极,参比电极采用的是Hg/HgO电极,辅助电极采用的是铂电极。首先在做电化学测试之前要对玻碳电极进行预处理,将准备好的玻碳电极润湿后搁置在0.5 um的Al2O3粉末上进行研磨抛光,之后用去离子水冲洗干净,再按次序将玻碳电极放置在装有乙醇、去离子水的烧杯里洗涤6~8 min,自然风干。称取一定量的制备好的样品于称量瓶中,然后用移液枪移取无水乙醇和一定浓度的Nafion溶液,摇匀以后超声成均一的墨水状溶液。然后用移液枪移取上述溶液逐滴地涂抹在已经洗涤干净的玻碳电极表面,自然风干。
本实验使用CHI 660E电化学工作站进行电化学测试,测试采用三电极二回路的测试体系。将粘附催化剂的玻碳电极作为工作电极,电极的有效面积是0.19625 cm2,辅助电极为铂电极,Hg/HgO电极作为参比电极,电解液为0.1 mol/L的KOH溶液,测试温度为室温。测试过程根据需要向电解液中通入氧气或氮气。
3. 结果与讨论
3.1. XRD分析
纯α-MnO2及X (Fe、Co、Cu)/α-MnO2的X射线衍射图谱见图1(a),从图中可以看出,不同元素掺杂所制备出水热产物的各衍射峰与纯氧化锰的粉末衍射标准谱(JCPDS No.44-0141,四方相α-MnO2,晶胞参数为a = b = 9.78 Å,c = 2.86 Å,空间群I4/m)很好的重合到了一起且没有其它单质元素或其化合物的产生,证明水热反应的产物是纯的α-MnO2 [15]。 为了进一步分析不同元素掺杂对氧化锰晶格的影响,某一晶面(211)衍射峰的局部放大见图1(b)。很明显的可以看到过渡金属元素掺杂后氧化锰的峰位均有一定程度的偏移,偏移的大小与所掺杂元素的种类有关。谢乐公式所计算出的数据见表1,通过谢乐公式计算可知,未掺杂其它元素的纯氧化锰晶粒最大,而掺杂Fe、Co、Cu后的氧化锰晶粒的尺寸均有一定程度的减小,这可能是由于其它元素的引入造成氧化锰晶体生长过程中有了更多的成核位点所导致。
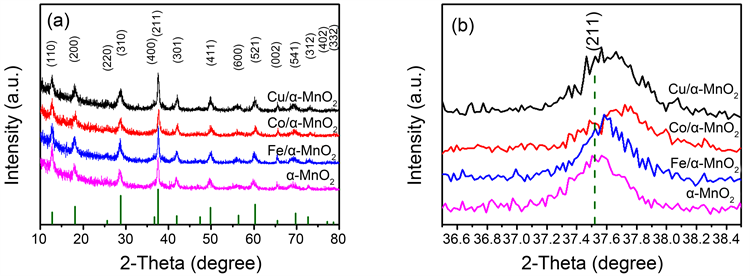
Figure 1. XRD patterns of the pristine α-MnO2 and X (Fe, Co, Cu)/α-MnO2
图1. 纯α-MnO2及X (Fe、Co、Cu)/α-MnO2的X射线衍射图谱

Table 1. Scherrer formula grain size calculated
表1. 谢乐公式所计算的晶粒尺寸大小
3.2. SEM分析
水热合成方法制备的纯α-MnO2及X (Fe、Co、Cu)/α-MnO2的扫描电子显微镜情况见图2,其中图2(a)为纯α-MnO2,图2(b)为Co/α-MnO2,图2(c)为Fe/α-MnO2,图2(d)为Cu/α-MnO2。尽管纯纯α-MnO2的纳米线略有团聚,但仍能够从图中看出水热法制备出的产物均为纳米线,未出现纳米球或纳米片等其它形貌。此外,从扫描电镜的图中可以看到掺杂其它金属的氧化锰纳米线整体是比未掺杂氧化锰纳米线的长径比要更加大一些,这可能与其中不同过渡金属离子掺杂进去后对氧化锰的生长过程造成相应的影响而导致。而在掺杂不同过渡金属元素后的氧化锰纳米线中,其长径比的大小由大到小分别为Co/α-MnO2 > Fe/α-MnO2 > Ni/α-MnO2。这些形貌上的差异可能会一定程度上影响其电催化性能。
 (a) α-MnO2
(a) α-MnO2  (b) Co/α-MnO2
(b) Co/α-MnO2  (c) Fe/α-MnO2
(c) Fe/α-MnO2  (d) Cu/α-MnO2
(d) Cu/α-MnO2
Figure 2. SEM images of the pristine α-MnO2 and X (Fe, Co, Cu)/α-MnO2
图2. 纯α-MnO2及X (Fe、Co、Cu) /α-MnO2的扫描电子显微镜图
3.3. CV分析
纯α-MnO2及X (Fe、Co、Cu)/α-MnO2的CV曲线见图3。从图中可以看到,所有的催化剂均出现了明显的还原峰,这说明采用上述掺杂方法所制备的催化剂都具有氧还原性能。此外,通过图3也可以判断出该反应为完全不可逆体系,由于电极反应的不可逆,逆反应非常迟缓,导致正向产物来不及发生反应就扩散到溶液内部之中,因此在循环伏安图中看不到反向扫描的电流峰。在各类元素掺杂的催化剂的对比中,Co/α-MnO2的峰位为678 mV,Fe/α-MnO2的峰位为662 mV,Cu/α-MnO2的峰位为628 mV,纯α-MnO2的峰位为526 mV。可以看到Co、Fe、Cu掺杂的α-MnO2催化剂峰位更正,而未掺杂的α-MnO2催化剂峰位则比掺杂后的各类氧化锰催化剂峰位至少小了100多个mV。峰位越正说明催化反应的过电位越低,催化活性越强。这证明了Fe、Co、Cu等过渡金属的掺杂对氧化锰催化剂的催化活性能够得到提高,而对于掺杂过渡金属元素的氧化锰所制备的催化剂来对比,Co/α-MnO2的峰位大于Fe/α-MnO2的峰位大于Cu/α-MnO2的峰位,说明Co的掺杂对于氧化锰的催化活性的提升可能大于Fe、Cu的掺杂。另外我们还可以从图中观察到按照Fe、Co、Cu掺杂的顺序,氧还原归属峰的峰高(峰电流值)也不断增大,峰电流越大表明氧还原反应的速率越快。因此从这角度来分析,Fe掺杂的催化剂有与Co掺杂接近的峰位,但其峰电流值更高,氧还原速率更快,Fe掺杂的催化剂对催化活性的提升可能大于Co、Cu的掺杂。

Figure 3. CV curves of the pristine α-MnO2 and X (Fe, Co, Cu)/α-MnO2
图3. 纯α-MnO2及X (Fe、Co、Cu)/ α-MnO2的CV曲线

Figure 4. CV curves of the pristine α-MnO2 and X (Fe, Co, Cu)/α-MnO2
图4. 纯α-MnO2及X (Fe、Co、Cu)/ α-MnO2的CV曲线
3.4. LSV分析
LSV测试是当下评价催化剂催化活性的重要指标,其中半波电位、起峰电位越正表明该催化剂所需要动力学条件越小,而极限电流密度越大则说明单位面积内该催化剂越能够帮助氧还原反应的进行,产生更多的电流。各种元素体相掺杂氧化锰的在1600 rpm下的LSV曲线见图4,图中涉及到20%商用铂碳,Ag/α-MnO2,X (Fe、Co、Cu)/α-MnO2以及α-MnO2催化剂。通过数据分析见表2,各类催化剂的起峰电位(极限电流密度值0.5%的电流密度所对应的电位)按照上述排序顺序逐渐减小,这说明了过渡金属(Fe、Co、Cu)的体相掺杂能够有效的提升氧化锰催化剂的氧还原活性。其中Fe/α-MnO2具有能够接近Ag/α-MnO2的能力,而Ag掺杂的催化剂具有与20%商用Pt/C接近的性能。从半波电位(极限电流密度值一半的电流密度所对应的电位)的角度来分析,其对应的结果与起峰电位的结果基本相似,同样可以证明这一论点。但从极限电流密度的数据中,Fe、Co、Cu掺杂的催化剂具有比商用铂碳更大的极限电流密度,这说明Fe、Co掺杂的催化剂虽然在起峰电位和半波电位上有短板,但更高的极限电流密度说明其具有更高的氧化还原速率,催化活性也能够与接近于Ag掺杂的催化剂。
Table 2. LSV comparative data of different catalysts at a scan rate of 1600 rpm
表2. 各类催化剂在1600 rpm扫描速率下的LSV对比数据
为了进一步证明其电催化效果,各类催化剂在400 rpm、625 rpm、900 rpm、1225 rpm、1600 rpm这五个不同转速下的LSV曲线以及依据该图数据进行转移电子数计算的K-L曲线见图5 [16] [17]。 随着转速的增大,O2的传质速率不断增加,进而有更多的氧气吸附在催化剂表面发生氧还原反应,因此各类催化剂的极限电流密度随着转速的增大而增大。从图中可以看出,在0.25 V、0.3 V、0.35 V、0.4 V这四个不同电位下依据K-L方程所拟合出来的直线几乎是平行的,具有良好的线性关系,意味着不同电势下的表观电子数n是一样的。通过计算可以得到20%商用铂碳,Ag/α-MnO2,X (Fe、Co、Cu)/α-MnO2以及α-MnO2的转移电子数分别是3.7、4.0、4.4、4.0、3.5、3.1 (由于转移电子数的结果是经过数据计算而非实验室直接测量,因此出现大于4的情况可能是由于计算中的各参数的变化导致误差,但本实验计算出的结果属于误差范围中),依据数据分析可以看出各类催化剂的均是按照接近四电子的反应方式来进行的。同时各类催化剂转移电子数的计算结果也与LSV中的分析结果想吻合,这说明过渡金属元素的体相掺杂的确能够提升氧化锰的催化效果,除去贵金属Ag以外Fe/α-MnO2提升的效果最佳。
3.5. EIS分析
为了更好的理解氧化锰催化剂的电极催化过程,采用旋转圆盘电极测试了不同交流催化剂的交流阻抗,测试溶液的浓度为0.1 mol/L KOH,测试的频率范围在10−2~105,测试结果见图6。

Figure 6. EIS curves of the pristine α-MnO2 and X (Fe, Co, Cu)/α-MnO2
图6. 不同元素X(Co、Fe、Cu)/α-MnO2的EIS曲线对比图
从图6中可以看出,氧化锰纳米线与各种过渡金属元素掺杂的氧化锰催化剂的阻抗图谱具有相似的形状,都是由一个位于第一象限内高频区的一个小圆弧连接着一个低频区的大圆弧所构成的,这说明在图中溶液界面与四种催化剂所发生氧化还原反应为同一过程。第一象限第二个圆弧的存在说明溶液界面与催化剂的反应时分布进行的,具体的进行过程如式 [18]:
(1)
(2)
图中O2得到一个电子转变为O− 2的过程反应在图7中是高频区所对应的小圆弧,而O− 2得到3个电子转变为OH−的过程在图6中为低频去对应的大圆弧。由于低频区圆弧的曲率半径远大于高频去圆弧,因此O− 2得到电子转变为OH−的过程是氧还原过程的速度控制步骤。
等效电路图是利用Zview软件模拟得出的,将图6中的交流阻抗数据进行了拟合,拟合的结果所体现内容见图7。从拟合结果来看,拟合曲线与实验数据很好的吻合在一起,这说明了拟合电路的合理性。
图7中每个元件都有其电化学上的意义。R1即Rs,代表的是电解液的电阻,R2、R3即Rct1、Rct2分别代表电极上两个电化学反应的电荷转移电阻,C代表的是第一个反应过程中的电容,CPE1代表用于修订非法拉第过程(包括双电层电阻)的恒定元件。其中,R2、R3的大小与电极反应的难以程度相关。同过对图7的数据进行拟合,所得到的R2、R3结果见表3。
Table 3. Actual parameters after fitting the components in the equivalent circuit diagram
表3. 等效电路图中个元件拟合后的实际参数
在电化学反应中,电荷转移电阻的大小影响着电化学反应的速度,阻值越小反应越容易进行。从表3中的R2值可以看出,四种催化剂的阻值均都较小且大小很接近,这说明这几类催化剂均能够有效的使O2得到一个电子转变为O− 2 (方程式1)。此外,对比表中的R3值能够看出,各催化剂的阻值很大并且数值相差都比较大,说明催化剂有O− 2转变为OH− (方程式2)的难度较大,是影响整个反应速率的控制步骤。通过数据的分析,可以很看出未掺杂的氧化锰催化剂比起过渡金属掺杂后的氧化锰阻值大了很多,催化反应的速率更慢,过渡金属的掺杂很明显的能够降低其反应电阻。而对其掺杂后的催化剂来说,Fe、Co、Cu的掺杂所带来阻值的影响仍然是按上述顺序依次减小的,这与CV和LSV分析的结果保持一致。
4. 结论
本文通过水热法将不同过渡金属元素硝酸盐中的过渡金属元素掺杂到氧化锰纳米线中,制备出了不同元素体相掺杂氧化锰的电催化剂。通过XRD、SEM等对催化剂成分和结构进行表征。此外,在上述的基础上,采用旋转圆盘电极装置对催化剂的电化学性能进行评估,测试了其CV、LSV、EIS等。得出的结论主要如下:
(1) 采用本文中所使用的水热反应方法能够成功制备出不同元素体相掺杂的氧化锰催化剂。并且各种过渡金属体相掺杂后的水热产物均为纯的氧化锰纳米线,没有其他的物相或晶体产生。
(2) 过渡金属元素掺杂后的氧化锰催化剂与未掺杂的纯氧化锰催化剂相比,其电催化性能均得到了不同程度上的提高,掺杂是能够提升催化剂催化活性的有效手段。
(3) 在不同过渡金属掺杂氧化锰催化剂的催化性能中,过渡金属Fe的掺杂能够带来最大的提升,Co次之,Cu掺杂的催化剂对于催化剂性能的提升最小
NOTES
*通讯作者。