1. 引言
近几年来,多模式成像方法由于可实现可视化治疗和提高诊断效果,获得快速发展,成为新研究热点。基于MRI与荧光成像各自的优势,以及弥补单成像模式的缺点,采用MRI和荧光双模式成像,在活体动物成像方面更具有优势,将之使用于诊断治疗中进行探索实验,并展望其潜在的临床与科研价值。
MRI和荧光的双模式成像,可用来定量测量在活体小动物体内荧光分子的三维分布 [1]。Fe3O4@CuInS2同时具备Fe3O4的磁性性质和CuInS2的荧光特性,可用于双模式成像。其中,磁性纳米颗粒表现出的超顺磁性和高饱和磁化现象受到各种领域的重视 [2] [3] [4],很多课题组提出不同合成方法,例如,超声化学合成、反胶束、微乳液、化学共沉淀、溶胶–凝胶、水热/溶剂热法等 [5] - [14]。为了更好地进入细胞和动物体内,需合成具有良好稳定性、分散性、无细胞毒性、水溶性良好的磁性纳米粒子,Fe3O4纳米粒子的粒径、形貌、磁能性和稳定性与合成的条件方法有紧密关系,本文通过溶剂热法方法合成的Fe3O4纳米粒子没有明显的团聚,且大小均匀,表现出良好的单分散性,可以稳定分散在水中。但是,Fe3O4纳米粒子由于具有磁性,容易沉聚,洗涤和重新分散较为困难,耐腐蚀能力差,且会吸收量子点中的能量而使荧光强度下降 [15]。因此,为了提高Fe3O4纳米粒子在极性溶液中的稳定性,不易沉聚,减少对量子点的能量吸收,对Fe3O4进行表面修饰是必不可少的。诸多研究课题组已经进行了一系列相关实验,其中Wang课题组在Fe3O4纳米粒子表面负载还原石墨烯(rGO),并修饰PEG,提高生物相容性,具有良好的光热治疗效果 [16];Chen等人利用PVBC-TMT修饰Fe3O4,增强对重金属离子的吸附,用于重金属离子的分离,对Pb2+的分离效果可以达到99%以上 [17];Shahin等人通过(3-氨基丙基)三乙氧基硅烷修饰Fe3O4纳米粒子表面,并利用甲壳胺和黄蓍胶进行包裹,构建化疗药物的载药体系,增强其pH和热敏感 [18]。
如图1所示,本实验采用溶剂热法Fe3O4纳米磁性粒子,在其表面修饰SiO2,形成Fe3O4@SiO2 纳米粒子。通过对Fe3O4@SiO2 纳米粒子表面修饰氨基活性基团,与经过修饰后带有羧基的CuInS2/CdS/ZnS量子点通过缩合反应结合,形成共价键,成功把量子点键合到磁性微球上,形成Fe3O4@CuInS2核壳结构,并将其应用于荧光成像和磁共振成像。
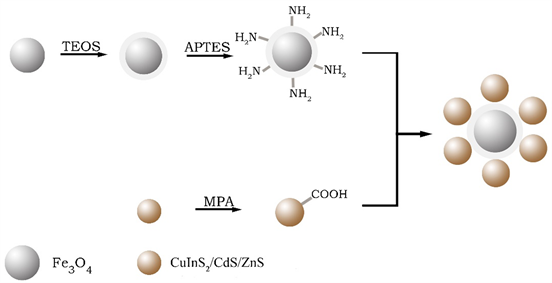
Figure 1. The preparation of Fe3O4@ CuInS2
图1. Fe3O4@ CuInS2制备过程
2. 实验部分
2.1. 主要试剂
柠檬酸三钠、三水合乙酸钠、磷酸氢二钠、氢氧化钠和正硅酸乙酯(分析纯,国药试剂),无水三氯化铁和氨丙基三甲氧基硅烷(上海麦克林),EDC(分析纯,上海生工),巯基丙酸(分析纯,北京伊诺凯科技有限公司),三氯化铟、谷胱甘肽和硫化钠(阿拉丁试剂)。
2.2. 仪器表征
利用X-射线粉末衍射仪(MiniFlex600,日本Riguka,以Cu Kɑ线进行扫描,扫描速度为0.02˚/0.01 s)研究材料的晶体结构;采用透射电子显微镜(Tecnai G2 F30 美国FEI公司)表征材料结构和形貌;采用荧光光谱仪(Cary Eclipse,安捷伦)研究量子点和复合材料的荧光性能,通过紫外可见光谱仪(UV-2600,日本岛津)表征材料光学性质,利用红外光谱仪(Nicolet Avatar 300,赛默飞世尔科技公司)表征材料的分子结构;通过磁滞回线测试仪(PPMS-9 Quantum Design公司)研究材料磁性性质;利用正置荧光显微镜(Axio Imager,德国蔡司)表征材料的荧光细胞成像,采用NMR分析仪(GYPNMR-10,上海寰彤)表征核磁共振细胞成像。
2.3. 实验方法
2.3.1. Fe3O4纳米颗粒的制备
准确称取0.6548 g无水三氯化铁,加入40 mL二乙二醇和1.00 g乙酸钠,充分搅拌,装入聚四氟乙烯反应釜中,在200℃烘箱中反应6 h。自然冷却至室温,离心,用水和乙醇混合溶剂(1:3)洗涤三次,除去多余二乙二醇和副产物,分散在40 mL去离子水中。
2.3.2. Fe3O4@SiO2的制备
利用经典的Stober法 [19],在Fe3O4纳米粒子表面包覆二氧化硅。取6 mL制得的Fe3O4分散液,加入100 mL三颈烧瓶中,依次往三颈烧瓶里加入6 mL水、50 mL无水乙醇超声10 min,开始机械搅拌加入2 mL氨水,然后缓慢注入0.2 mL TEOS,45℃水浴,机械搅拌6 h,最后得到Fe3O4@SiO2核壳材料。用水和无水乙醇混合液洗三次去除多余的乙醇和氨水,然后分散在20 mL水中形成灰黑色的Fe3O4@ SiO2分散液。
2.3.3. CuInS2量子点的制备
将0.01 M氯化铜(2 mL)、0.4 M柠檬酸钠(0.8 mL)、1 M氯化铟(0.08 mL)、0.0125 g谷胱甘肽加入40 mL去离子水中,混合均匀,继续加入1 M硫化钠(0.124 mL),95℃的水浴加热40 min;然后加入硫化镉2 mL (由0.04 M硫化钠、0.04 M乙酸镉各1 mL配制成),再40 min后加入硫化锌2 mL,每间隔45 min加入硫化锌2 mL (重复3遍),反应45 min后取出,放入EP管里备用。
2.3.4. Fe3O4@CuInS2的制备
为了使Fe3O4@SiO2能与量子点CuInS2/CdS/ZnS结合,在Fe3O4@SiO2表面修饰氨基使之与巯基丙酸修饰后的量子点CuInS2/CdS/ZnS-COOH脱水缩合形成稳定共价键结构。首先将制备好的Fe3O4@ SiO2水分散液用磁铁吸引去除水,加入40 mL无水乙醇,放置于三颈烧瓶中在剧烈机械搅拌下加入0.2 mL APTES,室温下反应2 h,反应完成用水和无水乙醇混合液洗三次,即制成Fe3O4@SiO2-NH2,保存在20 mL去离子水中备用。同时将制备好的CuInS2/CdS/ZnS量子点取20 mL于烧杯中,加入0.2 mL MPA后调pH至5.5,100℃水浴加热,磁力搅拌90 min后取出,加适量无水乙醇离心去除未反应的MPA,重新分散在20 mL去离子水中,获得CuInS2/CdS/ZnS-COOH分散液,加入8 mg EDC,避光活化30 min,获得活化完成的CuInS2/CdS/ZnS-COOH。最后,将制得的Fe3O4@ SiO2-NH2用磁铁吸引去除水分散在pH = 7的磷酸缓冲液中,与制得的活化完成的CuInS2/CdS/ZnS-COOH混合,避光反应12 h,反应结束后,用去离子水洗涤3次,分散在20 mL去离子水中即完成Fe3O4@CuInS2的制备。
2.3.5. 细胞成像
为了使Fe3O4@CuInS2能够更好的进入细胞,改善生物相容性,在Fe3O4@CuInS2纳米粒子表面修饰PEG。在Fe3O4@CuInS2的水分散液加入适量PEG,机械搅拌,室温反应2 h后,离心去除未连接的PEG。
采用生长对数期的宫颈癌细胞(Hela),以1 mL (含FBS的完全培养液)密度为10 W个/mL铺在每个培养皿。在37℃,5% CO2的细胞培养箱中培养至细胞密度达到50%~60%。移除培养基,加入1 mL不完全培养液,再加入40 μL材料,在37℃条件下培养2 h。PBS清洗3次后,用1 mL 2.5%戊二醛固定10 min,再次用PBS清洗3次,最后加1 mL PBS,于荧光显微镜下观察。
采用与细胞荧光成像相同的方法培育Hela,于NMR分析仪下观察。
3. 实验结果与讨论
3.1. 不同Fe3O4合成方法比较
分别比较两种方法合成Fe3O4纳米粒子,即沉淀法和溶剂热法。沉淀法采用FeCl3和FeCl2等体积混合后,加入过量的NH3·H2O作为沉淀剂,混合放入三颈烧瓶中,80℃水浴反应1 h。如图2所示,(a)为沉淀法所合成的磁性粒子,放置0.5 h后逐渐开始沉淀;放置24 h后,完全沉淀;(b) 为溶剂热法合成的Fe3O4纳米粒子,在水中形成分散均匀的体系,稳定性良好,保存半月仍未沉淀。
两种合成方法所制备的Fe3O4纳米粒子稳定性和分散性相差较大。根据Stokes定律可知,纳米粒子在水基体系发生表观沉降主要是由于纳米粒子之间发生团聚,所形成的团聚体越大,沉降越明显;而且沉降的速度与微粒半径以及微粒和分散介质的密度差的平方成正比。用沉淀法合成Fe3O4时,二价铁与三价铁的比例以及其他条件很难控制,极易被氧化,无法准确调控,会出现棕黑色颗粒。沉淀法所合成的Fe3O4磁性纳米粒子,粒径较大,易发生团聚,表观沉降速度加快,经过一段时间,粒子全部沉到容器底部,属于粗分散体系,该体系稳定性较差。用溶剂热法合成的Fe3O4磁性纳米粒子,合成条件可控,在水中分散较好,并且体系中布朗运动明显,粒子一方面受到重力作用而沉降,另一方面由于沉降使上、下部分的溶度发生变化,引起扩散运动,使浓度趋向于均匀,达到沉降平衡后,体系最下部浓度最大,随高度的上升浓度逐渐减小。

Figure 2. Fe3O4 synthesized by (a) chemical co-precipitation and (b) solvothermal method
图2. (a) 沉淀法和(b) 溶剂热法合成的Fe3O4
3.2. Fe3O4@CuInS2的结构分析
从图3可以发现,所制备Fe3O4磁性粒子呈现较强的结晶度,衍射峰位与Fe3O4标准图谱数据(JCPDS NO.79-0418)相吻合 [20]。Fe3O4@SiO2与纯Fe3O4相比,峰位基本一致,但峰强明显下降;而且在33˚附近出现较弱的峰,此为SiO2衍射峰(JCPDS NO.47-1300),证明SiO2成功包裹在Fe3O4表面。Fe3O4@CuInS2 核壳结构的XRD图显示Fe3O4主要特征衍射峰消失,在15-35°之间出现明显的宽峰,而CuInS2的第一特征衍射峰在27˚附近,结果表明CuInS2完整负载在Fe3O4表面。
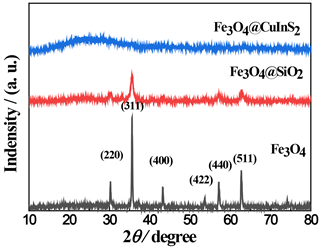
Figure 3. XRD images of different samples
图3. 不同样品的XRD谱图
图4(a)为Fe3O4磁性纳米粒子的TEM图,从图中可以看出Fe3O4为规则的球形,分散性较好,平均粒径约10 nm,有部分的Fe3O4磁性纳米粒子重叠,这是由于所合成的Fe3O4粒径较小,表面能较高,且为磁性材料,颗粒之间容易相互吸引。从插图可知,纳米粒子具有清晰的晶格条纹,说明所制备Fe3O4具有较好的结晶度,其晶面间距约为0.151 nm,对应Fe3O4的(511)晶面。从图4(b)中可看出SiO2较好的包裹Fe3O4,厚度较为均匀。图4(c)为CuInS2 /CdS/ZnS量子点的TEM图,从图中可发现,CuInS2/CdS/ZnS量子点分散性较好,粒径较均一。同时,具有比较明显的晶格条纹,晶格间约为0.302 nm,对应ZnS(111)晶面,表明成功制备出CuInS2 /CdS/ZnS核壳量子点。从图4(d)中可以发现,CuInS2/CdS/ZnS核壳量子点较好的分布在Fe3O4表面,证明成功制备Fe3O4@CuInS2核壳结构。

Figure 4. TEM image of (a) Fe3O4, (b) Fe3O4@SiO2, (c) CuInS2/CdS/ZnS QDs, (d) Fe3O4@CuInS2
图4. (a) Fe3O4磁性纳米粒子,(b) Fe3O4@SiO2,(c) CuInS2/CdS/ZnS 量子点,(d) Fe3O4@CuInS2核壳结构的TEM图
3.3. 光谱学和磁性分析
图5(a) 为激发波长为550 nm下的荧光光谱图,从图中可以看出,CuInS2/CdS/ZnS核壳量子点发射峰位于740 nm左右,荧光强度可达500左右,荧光效果良好。通过CuInS2表面结合羧基,优化CuInS2的荧光性能,得到的CuInS2/CdS/ZnS-COOH,发射峰仍位于740左右,荧光强度较CuInS2/CdS/ZnS核壳量子点明显增强,荧光强度可达550左右,结果证明CuInS2/CdS/ZnS修饰羧基有增强荧光的效果。复合后的Fe3O4 @ CuInS2在该峰位也出现发射峰,但是荧光强度明显降低,半峰宽也相较量子点增宽,可能是由于Fe3O4吸收了量子点的能量导致量子点荧光强度下降。
图5(b)为所制备样品的紫外可见吸收光谱,所有样品在可见光区均有吸收,由于纯Fe3O4为黑色,在全波段均有较强的吸收。
图5(c)为所制备样品的红外光谱。如图所示,Fe3O4纳米颗粒在568 cm−1有峰,对应为Fe3O4中Fe-O-Fe键伸缩振动的特征峰,在Fe3O4@SiO2中1091 cm−1附近的强而宽的吸收峰是对应Si-O-Si的不对称伸缩振动吸收峰,464、472 cm−1对应为Si-O-Si弯曲振动吸收峰,说明SiO2成功负载在Fe3O4表面。在1634 cm−1处的吸收峰对应C=O伸缩振动,3400~3500 cm−1处为磁性纳米颗粒表面羟基、氨基和吸附水的伸缩振动。在Fe3O4@CuInS2同时可见明显的Si-O-Si的不对称伸缩振动吸收峰,说明Fe3O4与CuInS2成功结合。
图5(d)为Fe3O4、Fe3O4@SiO2、Fe3O4@CuInS2的磁滞回线,结果表明,所合成的Fe3O4具有超顺磁性,磁化强度随着外磁场的增大而快速增加,在2 T的时候磁化强度达到饱和不再增加。合成的Fe3O4与Fe3O4@SiO2纳米粒子磁性曲线在1.5 T后达到饱和状态,说明都具有高饱和磁化强度 [21] [22] [23]。但是它们的磁矫顽力和剩余磁强度都为零,属于软磁范畴。Fe3O4@SiO2比饱和磁化强度(30 emu/g)小于Fe3O4纳米粒子(65 emu/g),说明Fe3O4经SiO2粒包覆导致磁性下降,同时表明硅有包覆上去,这与电镜结果相吻合。Fe3O4@CuInS2核壳结构的磁矫顽力和剩余磁强度仍为零,比饱和磁化强度有明显的降低,但仍具有超顺磁性,说明CuInS2/CdS/ZnS成功负载在Fe3O4@SiO2表面,形成Fe3O4@CuInS2核壳结构,与电镜结果相吻合,而且较低的磁性有利于在磁场中保持良好的分散性。
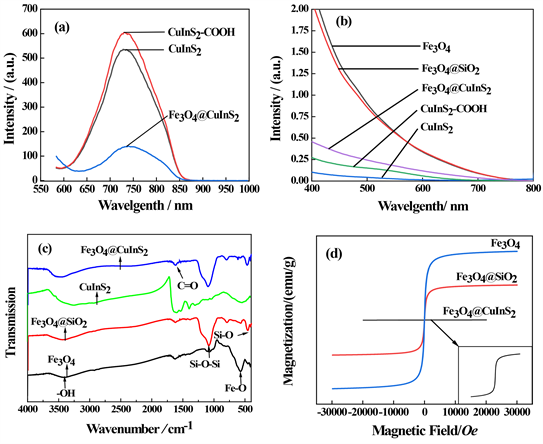
Figure 5. (a) Fluorescence spectra, (b) UV-vis absorption spectra, (c) infrared spectra, and (d) magnetic hysteresis loop of the as-prepared samples
图5. 所制备样品的(a) CuInS2的荧光光谱,(b) 紫外可见光区吸收光谱,(c) 红外光谱,(d) 磁滞回线
3.4. 细胞成像结果分析
将合成的纳米材料与密度合适的Hela细胞共同孵育,在荧光显微镜下定性观察细胞内部荧光物质的空间分布。图6为Fe3O4@CuInS2在Hela细胞中的荧光成像图,(a)为明场图,(b)为绿光激发下细胞成像图,(c)为叠加图。观察细胞荧光成像图,Fe3O4@CuInS2可有效地进入细胞,荧光强度较高,可以清晰反映细胞的形态,并且在绿光照射下具有明显可见的红色荧光,对细胞的损伤较小。这也进一步说明Fe3O4@CuInS2核壳结构可满足细胞荧光成像的要求。
将所合成的纳米材料与密度合适的Hela细胞共同孵育,在NMR分析仪下观察细胞磁共振成像效果。图7为Fe3O4@CuInS2在Hela细胞中的磁共振成像图,在T2加权成像下,空白组细胞显示高信号,含有PEG修饰的Fe3O4@CuInS2核壳结构的细胞显示低信号,空白组和实验组有明显可见的差别,表明Fe3O4@CuInS2核壳结构表现出良好的磁共振效果,也说明Fe3O4@CuInS2核壳结构可用作T2显影剂,可应用于磁共振/荧光双模式成像。
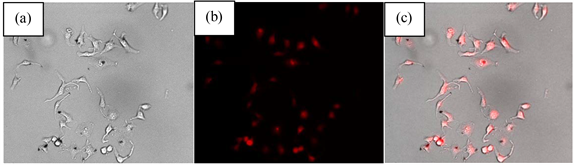
Figure 6. Fluorescence images of Hela cells treated with PEG-Fe3O4@CuInS2
图6. Fe3O4@CuInS2的荧光成像图

Figure 7. Magnetic resonance image of PEG-Fe3O4@CuInS2
图7. Fe3O4@CuInS2的磁共振成像图
4. 结论
本实验采用溶剂热法制备Fe3O4纳米粒子,所制的磁性纳米粒子没有明显的团聚,且大小均匀,表现出良好的分散性。而所制备CuInS2/CdS/ZnS量子点具有较强的近红外性能,且分散性好。采用改进的Stober法制备Fe3O4@SiO2磁性核壳结构,增强其生物相容性。并利用磁滞回线、光谱学、TEM和XRD等对磁性材料进行表征。结果表明,成功制得Fe3O4,具有较强的结晶度和顺磁性,SiO2有效包裹Fe3O4。通过酰胺缩合反应制得 Fe3O4@ CuInS2核壳结构,具有良好的磁性性质和磁感应强度,以及较强的荧光特性。利用表面PEG修饰,核壳结构能够有效进入细胞,展现出良好的近红外荧光成像和核磁成像效果,表明基于Fe3O4@ CuInS2核壳结构构建双模式成像系统的可行性。
基金项目
本项目研究由国家自然科学基金项目(51602053)、福建省自然科学基金项目(2019J01300)、福建省科技创新联合资金项目(2017Y9122)、福建省高等学校新世纪优秀人才支持项目(2018B031)、福建医科大学大学生创新创业训练资助项目(C18123)资助。
NOTES
*通讯作者。