1. 引言
乳腺癌是极为普遍的恶性肿瘤,国际癌症研究机构的调查结果显示,2018年环球女性发病率高达24.2%,发病率达到了女性恶性肿瘤第一位。临床研究证实,乳腺癌的分期不同,其对应的临床过程和治疗反应各异。乳腺癌分期的确定,为制定合适的治疗计划、估计患者的预后、协助评价治疗结果等提供了重要依据。
乳腺癌的分期方法各异,目前最通用的分期方法是TNM分期法 [1],由美国癌症研究会制定。TNM分期法取决于三个方面的表现:1) 癌肿的生长情况,以“T”来表示;2) 区域淋巴结的转移程度,以“N”表示;3) 远处脏器血行转移的有无,以“M”表示。这种分期方法基于病人的临床表现和肿瘤自身的发展程度。本文试图探寻从患者的基因表达数据的方向探寻其分期情况的方法。
目前机器学习方法有很多可以用来分类和预测,例如k-近邻(k-Nearest Neighbor, kNN) [2]、支持向量机 [3] (support vector machine, SVM)、随机森林 [4] 等方法,均可以对乳腺癌的分期进行很好的分类和预测。但存在一定的问题,如计算量较大、样本不平衡时结果较差等等。本文使用上述方法,对样本数据进行学习和预测。采用k-近邻方法对非过采样和过采样所得的样本进行测试,分别得到AUC值为0.598和0.781;采用支持向量机(SVM)分类法对两类样本测试,得到AUC的值分别为0.678和0.782;采用随机森林方法,得到的AUC的值分别为0.671和0.934。随机森林模型预测精确度高于其他两种,说明该模型对预测乳腺癌患者所处的分期具有较高的准确性。
为了充分考虑样本的平衡,本文对训练集的选取采用了过采样 [5] 的方法,使训练集成为平衡样本以提升随机森林的准确率和特异度及灵敏度。本文使用随机森林模型对过采样后的样本训练,得到预测准确率达96.75%,模型的灵敏性(sensitivity)和特异性(specificity)为97.5%和89.3%。对比随机森林模型对非过采样的训练后的结果,预测准确率有很大的提高。
2. 材料与方法
2.1. 数据材料
本文所用数据集选自TCGA数据库,基因表达数据包括1093个乳腺癌患者的基因表达数据,共18,004个基因。临床分期数包括1093个乳腺癌患者对应的分期,分期由stage I至stage V。
2.2. 数据预处理
2.2.1. 数据清洗
首先对数据进行去噪,删除分期缺失的数据;其次删除基因表达数据缺失值所占比例大于80%的数据,处理后得到1082个样本。将stage I,II的数据合并作为乳腺癌早期数据,stage III,IV,V合并作为晚期数据。
2.2.2. 构造Cohen’s d统计量
对每个基因计算其Cohen’s d值,并对基因进行降序排列,取排序前1000个基因作为后续的分类模型的特征。Cohen’s d值的定义见公式(1):
(1)
其中
表示第一类所有数据的均值,
表示第二类所有数据的均值;
、
分别表示第一类和第二类数据的个数;
、
表示第一类数据和第二类数据的样本方差。公式(1)中第一类、第二类指早期和晚期。
原数据有18,004个基因,对每个基因进行Cohen’s d值计算并进行降序排序,取前1000个基因。由上述的1082个样本的1000个基因组成的矩阵作为模型训练的样本数据集。
2.3. 构建训练集和测试集
2.3.1. 非过采样
采用非过采样的方法构建训练集和测试集,在整个样本数据集中随机抽取70%作为训练集,剩下的30%作为测试集。
2.3.2. 过采样
对早、晚期数据以9:1的比例构建训练集与测试集。由于所获数据中晚期数据只占总数据的26%,为不平衡数据且数据量适中,故选择过采样的方式构建平衡训练数据集 [5]。随机选取早期数据的90%和晚期数据的90%,分别得到722个样本和251个样本。有放回地在晚期数据251个样本中抽样,每次抽样后将样本放回原数据集,共抽取722次。最终得到早晚期占比一致的平衡样本集(共1444个样本)作为训练集。将剩下的10%早期数据(共81个)与10%晚期数据(共28个)作为测试集。
2.4. 预测模型
2.4.1. 随机森林模型
随机森林是一种基于数据驱动的非参数机器学习方法,结合了分类回归决策树和并行集成算法 [6]。首先从原始样本集中通过bootstrap重采样技术有放回地抽取样本子集作为训练集;在新的训练集的特征变量属性集中随机抽取若干个属性子集;从这个属性子集中选择最优属性进行节点分裂并形成分类树;M个随机树构成随机森林,分类结果也由这M个分类树投票决定。当新的测试样本输入随机森林模型中时,每一棵决策树都对样本投票,得票数最多的类别,即为样本的预测类别 [7]。本文在版本为3.5.3的R语言上进行实验,借用randomForest程序包 [8] 进行随机森林模型建立。
使用训练集对随机森林模型进行训练,得到反映其预测效果的混淆矩阵。训练后的随机森林模型对测试集进行测试,画出受试者工作特征曲线曲线ROC (receive operating characteristic curve),结合袋外错误率(OOB)来衡量预测的准确率。对于每一棵决策树,我们都可以得到一个OOB误差估计,将森林中所有决策树的OOB误差估计取平均,即可得到RF的泛化误差估计 [7]。袋外错误率定义见公式(2):
(2)
模型的灵敏度(真正类率),指真实类别为正类的样本中,分类预测也为正的比例,见公式(3):
(3)
其中TP表示真实类别为正,分类预测也为正的数目;FN表示真实类别为正,分类预测为负的数目。
特异度(真负类率),其定义为真实类别为负类的样本中,分类预测也为负的比例见公式(4):
(4)
其中TN表示真实类别为负,分类预测也为负的数目;FP表示真实类别为负,分类预测为正的数目。
2.4.2. k-近邻模型
k-近邻即kNN算法,kNN算法是典型的一种近邻分析方法。首先对所有数据进行训练集和测试集的划分。计算测试样本和训练集中每个样本之间的相似度,选择相似度最高的k个训练对象,根据这k个对象的类别对测试样本进行分类。其中相似度本文通过欧拉距离 [9] 来衡量见公式(5):
(5)
其中
是一个样本,上述表达式的值越小说明欧拉距离越小,两样本的相似度越大。本文使用R语言的kknn程序包建立k-近邻模型。
2.4.3. 支持向量机模型
支持向量机模型对输入变量与输出变量(二分类)之间的关系进行分析,进而对新样本的输出变量进行分类。模型以训练样本作为对象,将训练样本看做特征空间上的点,确定一个可将两类样本有效分离的超平面,即令平面两边的点距离平面间隔最大 [10]。本文使用R语言的e1071程序包建立SVM模型。
设γ为样本至分类平面的距离,ω是垂直于该平面的一个向量,则有公式(6):
(6)
进一步得到几何间隔公式(7):
(7)
目标函数为
。
2.5. 富集分析
根据随机森林中重要性得分,挑选出重要性排名前200的基因,并在metascape网站上进行富集分析,得到与这些基因相关的生物通路,并进行比对。
3. 结果
3.1. 随机森林模型结果
3.1.1. 非过采样结果
随机森林模型利用袋外数据建立了一个对误差的无偏估计 [11]。本文的非过采样样本集是由随机抽取70%样本组成训练集,剩余样本组成测试集。本文采用随机森林的袋外误差OOB和ROC曲线下的面积AUC作为评估随机森林的精确度的参数。表1和表2分别是随机森林模型对非过采样的训练集和测试集的混淆矩阵和袋外错判率。这里的I、II分别表示早期和晚期。

Table 1. Performance parameters of random forest training on non-oversampled training set
表1. 随机森林对非过采样训练集的性能参数
Table 2. Performance parameters of random forest training on non-oversampled test set
表2. 随机森林对非过采样测试集的性能参数
模型对非过采样的训练集和测试集的预测精度均不理想(表1和表2)。训练集袋外错判率36.75%,早期的预测错误率38.5%,晚期的预测错误率35%,错误率为预测错误的个数与总个数的比值;测试集中袋外错判率为38.65%,早期的预测错误率38.1%,晚期的预测错误率39.2%,灵敏度(TPR)和特异度(TNR)经计算分别为61.9%和60.8%。
ROC曲线 [12] 衡量了分类模型在任何数据集类别分布情况下的性能。本文采用ROC曲线评估算法衡量分类模型,对模型的性能进行评价。ROC以假正类率为横轴,以真正类率为纵轴,分别表示实际负类中被预测错误的比例和实际正类中被预测正确的比例。本文将病情早期定义为正类,晚期定义为负类进行研究。ROC曲线下面积AUC,其值是介于0.0到1.0之间的概率值,结合ROC曲线的形态可以直观定量的评价预测模型的好坏,AUC = 0.5代表分类器类似于随机猜测,没有预测价值,AUC = 1则代表一个完美分类器 [12]。
非过采样得到的AUC值为0.671 (图1),结果并不理想。通过分析认为,由于早期样本数据和晚期样本数据在数量上有很大差距,不平衡样本造成预测的准确率不理想。
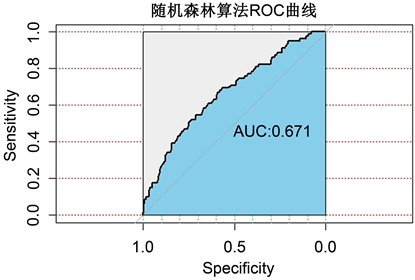
Figure 1. Roc curve of random forest algorithm (non oversampling)
图1. 非过采样时随机森林算法ROC曲线
3.1.2. 过采样结果
对晚期数据训练集进行重采样处理 [13],使其与训练集中的早期样本个数相同,通过随机森林模型对训练集进行预测,得到混淆矩阵见表3:
Table 3. Training set confusion matrix
表3. 训练集混淆矩阵
对过采样样本集进行学习,袋外错误率只有3.25%,小于非过采样训练集的袋外错误率36.75%,过采样样本预测准确率达到96.75%。
对测试集进行预测,得到混淆矩阵见表4:
Table 4. Test set confusion matrix
表4. 测试集混淆矩阵
随机森林模型对过采样的测试集的预测准确率为93.4%,灵敏度(TPR)和特异度(TNR)分别为97.5%和89.3%,均高于非过采样的测试集的结果。
模型对过采样样本训练得到的AUC值为0.934见图2,大于非过采样得到的AUC值0.671。因此我们认为过采样做训练集构建的模型更精准。
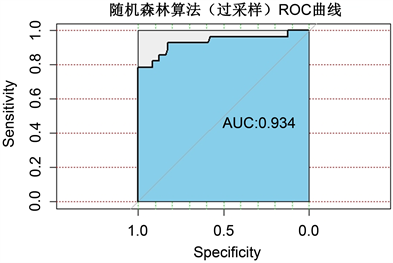
Figure 2. Roc curve of random forest algorithm (oversampling)
图2. 过采样时随机森林算法ROC曲线
3.2. 三种模型结果的比较
本文使用k-近邻、支持向量机和随机森林模型对非过采样与过采样的训练集进行训练。见表5,针对非过采样样本,支持向量机和随机森林的准确度较高于k-近邻模型,针对过采样样本,随机森林模型的准确度明显高于其他两类模型。综合以上情况,选择随机森林模型作为预测模型。
Table 5. AUC values of the three models
表5. 三种模型AUC值
3.3. 结果验证
本文采用十折交叉验证的方法来测试随机森林模型的准确性。将所有样本划分为10个数据集,不重复地选取其中一个数据集作为测试集,其他9个数据集作为训练集,并重复10次。保证每个数据集都被利用,以降低泛化误差。最终结果取十次交叉验证准确率的平均值。经过计算得到随机森林模型十折交叉验证平均准确率为96.71%。
4. 生物通路与富集分析
由于原数据集中的基因作用未知,以本文选用的基因表达数据预测分期是否有意义未知。为了进一步验证随机森林模型对于原数据集中基因表达数据的分期预测可行性,本文对输入特征(基因)进行了富集分析,并根据基因的生物学通路对其可行性进行判断。
4.1. 数据预处理
输入随机森林模型的数据集包含1000个基因。随机森林模型中的importance参数可以得到输入变量重要性测度矩阵,按照Mean Decrease Accuracy对基因降序排序,获取排序前200的基因。
4.2. 富集分析
将预处理得到的200个基因,使用metascape网站对这些基因进行富集分析。富集分析可以解释基因的功能或它在疾病发病中的作用,对基因的功能进行生物学解释 [14]。将数据输入可得到富集后的生物通路如下图3:
横坐标的值−log10(p)中的p为p-values。排序越靠前,−log10(p)值越大即p-values值越小,富集越显著,其颜色越深。
4.3. 生物通路的注释分析
上图3中富集较为显著且与癌症相关的生物通路为:氨基酸及其衍生物的代谢、依赖于P53蛋白的内源性凋亡信号通路以及胞浆钙离子浓度的正调节。
氨基酸及其衍生物的代谢。氨基酸不仅是构成蛋白质的基本单位,与肿瘤细胞也有密切的关系。有临床研究发现,乳腺癌患者血浆中蛋氨酸含量与正常人相比显著升高,异亮氨酸、亮氨酸、缬氨酸、精氨酸、赖氨酸、酪氨酸、苯丙氨酸含量与正常人相比显著下降 [15]。
依赖于P53蛋白的内源性凋亡信号通路。正常情况下,细胞中p53蛋白的含量极低;当细胞处于应激或受损伤的状态时,在某种异常信号的刺激下时,p53蛋白含量会迅速增加,阻止细胞恶性增殖 [16]。P53蛋白的作用有:介导细胞周期阻滞、参与DNA损伤的修复、和调节细胞的分化和衰老并抑制肿瘤血管增生 [16]。
胞浆钙离子浓度的正调节。肿瘤细胞中的Ga离子浓度远高于正常细胞:转化细胞内钙结合蛋白质/钙调蛋白含量比正常细胞多2倍。Ga离子启动的一些信号通路又能增加胞内Ga离子,连续启动Ga离子信号通路,使钙通道病理性地开放,导致细胞内Ga离子浓度长期居高不下,使多条信号通路不停地运转,此可称为Ga离子启动的环形信号转导通路,它能影响到细胞的机能、代谢和生存环境,并可能因此启动细胞的癌变过程 [17]。
由随机森林模型得到的200个基因获取的生物通路与癌症相关度较高。由此我们可以得出结论:对本文使用的基因表达数据使用随机森林模型进行分期预测是可行的。
致谢
在论文付梓之际,我们十分感谢项目的指导老师——陈园园副教授,项目的完成离不开她的耐心指导与悉心关怀。陈老师善于点亮我们的灵感、开拓我们的思维;项目遇到瓶颈时,她总会和我们一起分析问题出现的原因,并提出实用的方法与建议。陈老师不放过任何一个细微的错误,她严谨求实、一丝不苟的作风深深地影响着我们,使我们受益匪浅。最后感谢项目组的成员们,集体的智慧、齐心协力的精神与每个人的无私付出是保证项目成功的必要条件。
项目资金
本文研究工作由南京农业大学大学生研究训练计划项目(1923A13)提供资助。
NOTES
*通讯作者。