1. 引言
集成电路与LCD生产工艺常使用TMAH作为显影剂,TMAH是一种具有毒性的含氮有机物质,该类有机废水具有强碱性及高氮特性。目前中国大陆尚未有法规管制TMAH排放浓度,但由于近年来环保意识高涨,台湾环保署增订光电、科学园区污水下水道系统和晶圆制造业的放流水标准,新竹科学园区管理局也修改相关水质标准,将TMAH纳入二阶段管制(TMAH < 30 mg∙L−1)。
氢氧化四甲基铵分子式为(CH3)4NOH,结构式如图1,其分子量为91.15,是一种离子键结的物质,具有类似铵类(Amine)气味、极易溶于水、pH值大于13,水解后会形成OH−基、TMA+基(王及陈,2008) [1]。
本研究主要是在好氧条件下以生物分解废水中的TMAH。甲烷氧化菌分解四甲基铵盐类的代谢途径,主要是以连续氧化反应将四甲基铵依序降解成三甲基胺(Trimethylamine)、二甲基胺(Dimethylamine)、甲基胺(Methylamine),而伴随着降解过程,甲醛(HCHO)与氨氮会被释放出来(Anthony, 1982) [2]。

Figure 2. Mechanism metabolic pathway of TMAH
图2. TMAH的生物代谢途径
生物除氮过程包括生物发生硝化反应的好氧阶段和短时间缺氧过程,以提供生物的反硝化。用NH4-N的氧化和NO3-N、NO2-N的还原将氨氮转化成氮气,以达到去除氨氮的目的 [3]。根据以上相关文献汇整TMAH生物代谢途径如图2。
Lei等人 [4] 研究了一種在好氧和厭氧條件下處理包含TMAH、二甲基亞砜(DMSO)和單乙醇胺(MEA)的廢水,於兩種情況下都可能降解TMAH。另外,文獻顯示有多種可以分解TMAH之微生物,Mycobacterium sp.、Pseudomonas aminovorans (Urakami et al., 1990; Ghisalba et al. 1985) [5] [6] 可於好氧的環境下分解TMAH。厭氧條件下,Methanomethylovorans spp.、Methanococcoide spp.和Methanosarcina spp.具有直接降解TMAH的潛力 [7]。
水体中微生物是水域生态系统的重要组成部分,微生物群落状况与水质状况有着密切的联系,其结构的变化对水质污染负荷积累有很好的响应 [8] [9],它们在生源要素循环、有机物分解和污染物净化等方面起着非常重要的作用。近年来,在微生物群落多样性研究中倾向于采用分子生物学技术 [10] [11] [12],分子生物学技术是以细菌基因序列信息做为基础,通过细菌的基因序列来探讨样品中的微生物的组成及变化,由于其不需要对样品中的微生物进行分离培养,就能够快速检测出大量未培养的微生物,与传统研究方法相比具有重现性高、节省时间等优点 [13] [14]。而16S rDNA通常作为鉴定生物物种的特征核酸序列,16S rDNA是进行高通量测序和菌种鉴别 [15] [16]。本研究主要采用Illumina MiSeq测序技术研究不同TMAH浓度下对硝化系统微生物群落结构差异组成。
2. 实验与方法
2.1. 污泥来源与采集
本实验所使用的硝化污泥来源于台湾某半导体厂内氨氮生物处理槽体,批次反应器内进水是利用RO水人工配制氨氮与TMAH的混合废水。批次反应器内初期先添加NH4Cl使氨氮浓度为200~400 mg∙L−1左右,硝酸盐氮及亚硝酸盐氮趋近于0,实验期间pH控制于7.5~8.5。
反应稳定期间,每日取样分别分析氨氮、硝酸盐氮、亚硝酸盐氮及总氮之数值。表1为生物槽体内的初始与最終数值。

Table 1. The initial and final concentration of wastewater quality
表1. 各组废水的初始与最终组成分
单位:mg∙L−1。
2.2. 分析方法
本研究之水质分析为监测pH、NO2-N、NO3-N、NH3-N、TN之变化与并利用TN与氨氮换算成TMAH浓度进而观察TMAH的降解情况,表2为本研究水质之分析方法。
Table 2. Methods of analysis for wastewater quality
表2. 水质之参考分析方法
2.3. 微生物核酸萃取、16S rRNA基因测序库(Amplicon Library)建立与高通量測序
反应器运行16天后,取样送至Genomics (Taiwan)公司进行微生物核酸与萃取测序,此部分分析将采用次世代测序技术Illumina MiSeq 2000 sequencing system (San Diego, USA)。
3. 结果与讨论
3.1. 样品测序质量分析
稀释曲线(Rarefaction Curve)用于描述组内样品多样性的曲线,可直接反映测序数据量的合理性,并间接反映样品中物种的丰富程度。对样品随机抽样,以抽取的测序数据量与对应的物种数构建稀释曲线如图3所示,当随机抽取的测序数量大于30000条时,曲线趋向平坦,表明测序数据量渐进合理,更多的资料量对发现新的OUT (Operational Taxonomic Units)边际贡献率较小。而当测序数量相同时,细菌群落OTU数量表现为组别B (TMAH 1000 mg∙L−1) > 组别A (TMAH 0 mg∙L−1) > 组别C (TMAH 3000 mg∙L−1),表明组别B (TMAH 1000 mg∙L−1)微生物丰富度明显高于其他样品。
Rank Abundance曲线(图4)反映了样品中物种的丰富度和均匀度。当水平曲线跨度较大,表示物种的丰富度较高;而在垂直方向上曲线平滑程度较小,表明测定样品中物种的均匀程度相对较低。图4明显得知组别B-TMAH 1000 mg∙L−1的曲线跨度大,代表物种的丰富度高于其他两组。
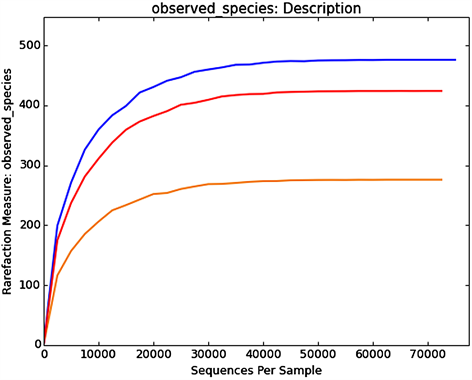
Figure 3. Rarefaction curve for Sample-A, B, C
图3. 样品的稀释曲线图
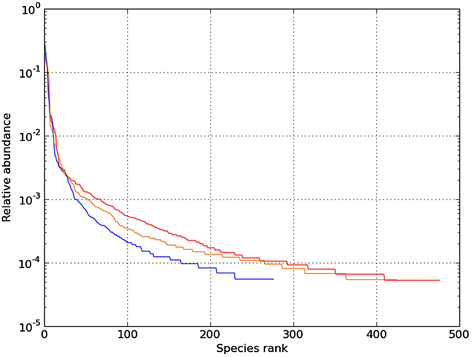
Figure 4. Rank Abundance curve for Sample-A, B, C
图4. 样品的Rank Abundance曲线图
3.2. 细菌菌种丰度与歧异度
由表3可知,组别A-TMAH 0 mg∙L−1得到的拼接序列数共有76,832条Tags,在97%的序列相似性条件下,可分为2809个OTUs,包括40,260条Unique Tags,40,247条Taxon Tags;组别B TMAH-1000 mg∙L−1得到的拼接序列数共有78,493条Tags,在97%的序列相似性条件下,可分为2651个OTUs,包括43,352条Unique Tags,43,335条Taxon Tags;组别C-TMAH 3000 mg∙L−1得到的拼接序列数共有74,489条Tags,在97%的序列相似性条件下,可分为1851个OTUs,包括38,180条Unique Tags,38,180条Taxon Tags。从这些资料可以看出,生物硝化系统中含有TMAH 1000 mg∙L−1者,其与TMAH 0 mg∙L−1者的生物菌群数量差异不大,但丰富度略高于TMAH 0 mg∙L−1者;而当TMAH提高至3000 mg∙L−1总体微生物总数变低且微生物种类比较单一。TMAH的加入使得环境内的菌种丰富度提高,但浓度提高到一定比例时,TMAH的毒性会造成微生物数量及种类变少。
Table 3. Numbers of Tags and OTUs
表3. 样品的Tags和OTUs数目统计
样品中的OTU数量可代表物种丰富度,可进一步分析其物种丰富度指数(Community Richness)、物种均匀度指数(Community Evenness)以及物种多样性指数(Community Diversity)。物种丰富度即为物种的数目,数目愈多代表丰富度愈大,生物多样性也就越高。物种均匀度说明一个群集中各个物种个体数目的分配状况,反映出各个物种个体数目分配的均匀程度,一个群集中各物种的个体数目愈相近者,代表物种均匀度高。在物种丰富度相同的状况下物种均匀度越高者,则说明物种多样性越高。
根据OTU聚类分析结果和研究需求,当样品数大于5时,对所有样品进行均一化处理,分析不同样品之间共有和特有的OTU并绘制花瓣图(图5)。从图中可以直观的看出不同样品OTU数目组成相似性重迭情况,3个样品共有的OTU数目为105,不同样品中特有OTU表现为组别B-TMAH 1000 mg∙L−1最高(155),组别A-TMAH 0 mg∙L−1次之(121)组别C-TMAH 3000 mg∙L−1最低(57)。

Figure 5. Flower diagram based on OTUs
图5. 基于OTUs绘制的花瓣图
3.3. TMAH浓度对微生物群落结构之影响
稀释曲组别A (TMAH 0 mg∙L−1)序列数据库比对后共得到14个门、26个纲、51个目、71个科、113个
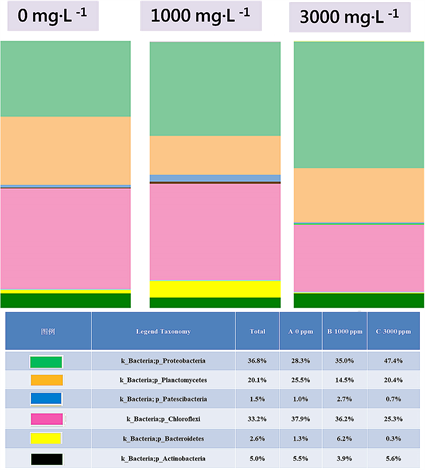
Figure 6. Relative abundance of OTUs assigned at the phylum level
图6. 分类层级为门之结构组成
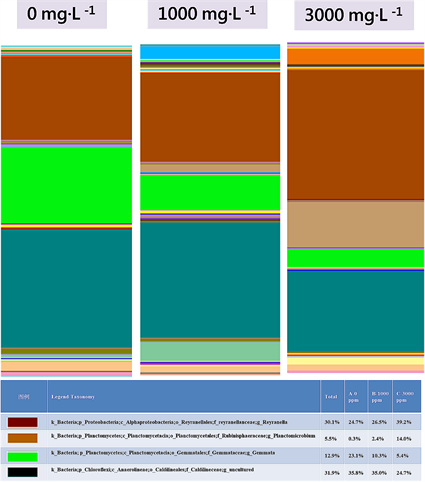
Figure 7. Relative abundance of OTUs assigned at the genus level
图7. 分类层级为属之结构组成
属及113个种。以门分类层级来看,Chloroflexi为最优势、Proteobacteria次之再来是Planctomycetes (图6)。以属的分类层级来看,Caldilineaceae (Genus uncultured)为最优势、Reyranella次之再来是Gemmata (图7)。
组别B (TMAH 1000 mg∙L−1)序列数据库比对后共得到18个门、32个纲、65个目、96个科、150个属及175个种。以门分类层级来看,Chloroflexi为最优势、Proteobacteria次之再来是 Planctomycetes (图6)。以属的分类层级来看,Caldilineaceae (Genus uncultured)为最优势、Reyranella次之再来是Gemmata (图7)。
组别C (TMAH 3000 mg∙L−1)序列数据库比对后共得到14个门、23个纲、46个目、62个科、97个属及113个种。以门分类层级来看,Proteobacteria为最优势、Chloroflexi次之再来是Planctomycetes (图6)。以属的分类层级来看,Reyranella为最优势、Caldilineaceae (Genus uncultured)次之再来是Planctomicrobium (图7)。
3.4. 样品复杂度分析
Alpha多样性主要是探讨样品内的微生物群落多样性,通过分析评估各样品中微生物群落的物种丰富度和多样性的差异。序列相似度大于97%的情况下可聚类成一个OUT,通常被认为是源自同一个种(Species Boundary),所得的Alpha多样性指数取平均值如表4所示。分析表明,所有取样点的多样性测序结果Coverage均高于99%,表明测序结果覆盖度好且具有较高可信度,能够代表样本的真实情况。Shannon指数和Simpson指数则是反映物种丰富度和均匀度的2个重要参数,其值越大代表个体分配越均匀、群落多样性越高,从表4可以看出,3组菌落的Shannon多样性指数和Simpson多样性指数排序分别为组别B (TMAH 1000 mg∙L−1) > 组别A (TMAH 0 mg∙L−1) > 组别C (TMAH 3000 mg∙L−1),结果表明高浓度TMAH系统中细菌微生物群落多样性相对较低。Chao1指数是生态学中用于估算样本物种丰富度的常用指数,表现为组别B (TMAH 1000 mg∙L−1)值最大、组别C (TMAH 3000 mg∙L−1)最低,表明前者群落的丰富度最高,而后者丰富度最低。
Table 4. Diversity index of bacterial community
表4. 微生物群落多样性指数
3.5. 聚类分析
为了进一步得到不同样品间群落结构差异,对OTU进行多序列比对并基于Weighted Unifrac距离矩阵构建非加权组平均聚类树形图(Unweighted Pairgroup Method with Arithmetic Mean, UPGMA)结果如图8所示,组别B (TMAH 1000 mg∙L−1)和组别A (TMAH 0 mg∙L−1)相似度最高,组别C (TMAH 3000 mg∙L−1)与其他样品间距离最远,相似度最低。
3.6. 综合讨论
由表5(A)以上的分析结果可知,浮霉菌门(Planctomycetes)中的Gemmata属于组别A占22.52%,在组别B及组别C却减少至10.29%及5.13%;放线菌门(Actinobacteria)中的Rhodococcus属于组别A占2.68%,在组别B及组别C也减少至 < 2%,相对数量明显,表明TMAH对Gemmata属及Rhodococcus属有较大的抑制作用。
表5(B)显示组别B (TMAH 1000 mg∙L−1)与组别A (TMAH 0 mg∙L−1)相比,增加TMAH浓度的变异性,带入了新的真菌物种(如:变形菌门(Proteobacteria) Brachymonas属、拟杆菌门(Bacteroidetes) Flavobacterium属),其菌种丰富度也明显提高。但以上菌种于TMAH浓度3000 mg∙L−1时,其占比锐减,受到高浓度TMAH一定程度的抑制。
变形菌门(Proteobacteria)中的Reyranella属通常出现在含氮系统的中后期,相关研究表明,变形菌门在生物脱氮及诸多污染物降解过程中起着重要作用 [12],随着TMAH浓度增加其生长环境和代谢基质方面都更加有利于Reyranella属繁殖,最终表现为随着TMAH浓度的增加其数量也增加,以上现象于浮霉菌门(Planctomycetes) Planctomicrobium属及变形菌门(Proteobacteria) Castellaniella属的占比中也呈现递增的趋势。
Table 5. (A) TMAH inhibition (B) relative abundance of TMAH 1000 ppm (C) optimum bacteria of high TMAH concentraion
表5. 各项目探讨(A) TMAH抑制(B) TMAH1000相对丰富度(C)高浓度TMAH最适菌
4. 讨论
综合以上各条件实验之结果,本研究初步之结论如下:
1) 从各组细菌物种分类可以看出,硝化系统中的绝对优势的细菌主要来自变形菌门(Proteobacteria)、浮霉菌门(Planctomycetes)、绿弯菌门(Chloroflexi)。
2) 组别A TMAH 0 mg∙L−1呈现良好的硝化反应,其属之结构组成中浮霉菌门(Planctomycetes) Gemmat属的比例比其他两组高。组别C TMAH 3000 mg∙L−1高浓度下变形菌门(Proteobacteria) Reyranella属明显比组别A及组别B高。
3) 细菌丰富度表现为组别B (TMAH 1000 mg∙L−1) > 组别A (TMAH 0 mg∙L−1) > 组别C (TMAH 3000 mg∙L−1)。组别B (TMAH 1000 mg∙L−1)与组别A (TMAH 0 mg∙L−1)相比,增加TMAH浓度的变异性,可能带入了新的真菌物种,其菌种丰富度也明显提高。
4) 细菌群落OTU数量表现为组别A (TMAH 0 mg∙L−1) > 组别B (TMAH 1000 mg∙L−1) > 组别C (TMAH 3000 mg∙L−1)。测试期间TMAH 3000 mg∙L−1菌种分布受到诸多因素(TMAH浓度高、硝化作用抑制)的综合制约影响,和其他样品距离较远,物种总量和丰度具有特异性。