1. 前言
谷氨酸棒杆菌(Corynebacterium glutamicum)在1957年首次被发现是作为谷氨酸的天然生产者 [1]。因其具有生长迅速 [2],基因组稳定 [3],代谢网络具有可塑性 [4] 等优点,谷氨酸棒杆菌成为了重要的工业微生物。研究作为基因转录调控元件的谷氨酸棒杆菌启动子具有重要的理论和应用价值。启动子活性的强弱在很大程度上影响着转录的起始、持续作用的时间以及外源基因的表达水平,进而宿主细胞中外源基因的表达程度也受到了影响。基因翻译区的上游区域是属于启动子序列的区域,通过其与对应的转录因子互作,可以精确调控下游基因的转录 [5]。
细菌启动子活性主要取决于启动子与一致序列的相似性,其活性会被−10区、−35区以及一些在致序列附近的非高度保守核苷酸所影响 [6]。研究表明,在−10区中心的TATAAT序列会增强启动子的活性,而−10-六聚体上游的TG二聚体则对启动子的强度有影响 [7]。且有研究数据表明,对启动子−10区和−35区的碱基进行突变会对启动子的转录水平造成较大影响,而改变−10区和−35区之间的碱基序列则对启动子的活性影响较小 [8]。谷氨酸棒杆菌中的内源启动子可通过以下3种主要的方法寻找:1) 基于随机序列合成启动子库,2) 启动子突变和3) 使用启动子预测软件,基于其转录组、基因组或蛋白质组来鉴定新的高效的启动子 [9]。值得注意的是,谷氨酸棒杆菌中启动子的−10区的碱基序列是高度保守的,-35区则相对保守 [10]。尽管大肠杆菌中的启动子P-tac和LacZ均被证明能在谷氨酸棒杆菌中使用,但低效率与泄露表达使这些外来启动子不能完美的表达目的蛋白 [11]。因此,选择高活性的内源启动子成为一大热点。然而关于谷氨酸棒杆菌高效内源启动子鉴定的相关报道很少,这限制了应用谷氨酸棒杆菌细胞工厂高效生产重组蛋白的研究。本研究望通过分析在谷氨酸棒杆菌细胞全蛋白二维电泳图上展现的高表达蛋白斑点,以鉴定谷氨酸棒杆菌内源强启动子序列,为构建高效的谷氨酸棒杆菌细胞工厂提供有力的基因表达元件。
2. 材料与方法
2.1. 实验材料及培养基
大肠杆菌E. coli BL21 (DE3);野生型谷氨酸棒杆菌C. glutamicum ATCC13032为本研究所用到的菌株。E. coli-C. glutamicum启动子穿梭载体为实验室前期构建的载体 [12]。表1为本研究中所用到的引物。
称取5 g的Yeast Extract、10 g的Tryptone以及10 g的NaCl配制成1 L的LB培养基(pH 7.0),用于大肠杆菌菌株培养,培养温度为37℃;本试验还需用到LBHI培养基(pH 7.0),配制1 L该培养基需称取5 g的Tryptone 、2.5 g的Yeast Extract、5 g的NaCl和18.5 g的脑心浸液,此培养基用于谷氨酸棒杆菌菌株的培养,培养温度为30℃。

Table 1. Primers used in this study
表1. 研究中使用的引物
2.2. 前期工作
在实验室前期工作中,获取了谷氨酸棒杆菌ATCC13032的细胞全蛋白,并在相应公司将获得的细胞沉淀进行二维电泳及质谱分析。公司返还相关数据后,利用质谱法分析我们从二维电泳图上选取的一些染色较深的蛋白点,同时使用NCBI网站查询其氨基酸序列。最后结合谷氨酸棒杆菌ATCC13032全基因组注释信息对二维电泳图上的高表达蛋白斑点进行分析,分析所含蛋白质的等电点、分子量等相关数据,以便后续与二维电泳图上的蛋白斑点进行比对。同样,我们在前期工作中在启动子预测网站(http://www.cbs.dtu.dk/services/Promoter/)上对编码人为选择的染色较深的蛋白点基因进行了启动子分析,选择的区域为起始密码子上游的DNA序列处,获得了一系列预测的启动子片段。
2.3. 扩增目的片段
为扩增目的启动子的DNA片段,采用寡核苷酸退火法将设计好的正反引物合成双链DNA。引物序列atatggatccgtaacgtggcaaaacgaacaatgtctcactagactaaagtgagatcgacaaagcttaatt (P1937-F)和 aattaagctttgtcgatctcactttagtctagtgagacattgttcgttttgcctcgttacggatccatat (P1937-R)用于合成启动子P1937片段,利用引物序列atataagcttaattgttatccgctcacaattccacacattatacgagccgatgattaattgtcaacagctcaggatccaatt (Ptac-M-F)及aattggatcctgagctgttgacaattaatcatcggctcgtataatgtgtggaattgtgagcggataacaattaagcttatat (Ptac-M-R)扩增启动子Ptac-M片段。正、反向引物各1 μL囊括在制备的50 μL PCR反应体系。经过10倍稀释的正反引物及所需的其他试剂充分混匀后进行PCR反应,反应条件同张献等的方法 [13]。
2.4. 构建重组载体
启动子探测载体上含有的BamHI及HindIII的酶切位点,因此在对引物进行设计时也加入这两种酶的酶切位点,利用这两种限制性内切酶分别对启动子P1937和Ptac-M以及启动子探测载体pDXW-11在相应酶切位点处进行双酶切,随后T4连接酶的使用,能让片段用与启动子探测载体相连接形成重组载体,后通过热激转化法转化进E. coli感受态细胞中。其中E. coli感受态细胞的制备以及转化方法遵循Sambrook等的方法进行 [14]。转化并利用氯霉素抗性验证成功后,即获得了携带启动子P1937的重组载体pDXW-11-P1937和携带启动子Ptac-M的重组载体pDXW-11-Ptac-M。
2.5. 谷氨酸棒杆菌电转化
首先遵循Xu等 [15] 的方法制得谷氨酸棒杆菌感受态细胞。后通过电击转化方式将重组探测载体pDXW-11-P1937和pDXW-11-Ptac-M转化进C. glutamicum ATCC13032感受态细胞中,其转化方法同样依据Xu等 [15] 的方法进行。利用氯霉素抗性验证成功后,即构建了带有启动子P1937的重组菌株C. glutamicum/pDXW-11-P1937和带有启动子Ptac-M的重组菌株C. glutamicum/pDXW-11-Ptac-M。
2.6. 谷氨酸棒杆菌内源启动子活性分析
2.6.1. 耐受性实验
载体上存在氯霉素筛选标记,因此制备具有不同氯霉素浓度梯度的LBHI平板,并将携带有启动子P1937的谷氨酸棒杆菌ATCC13032和携带启动子Ptac-M的谷氨酸棒杆菌ATCC13032这两种菌株在以上平板上进行接种,并观察菌落生长情况,初步判断启动子活性强弱。
2.6.2. 氯霉素乙酰基转移酶酶活检测
用100 mmol/L Tris-HCl (pH 7.8)溶液洗涤收集到离心管中的30 mL培养液的细胞沉淀,高速离心后,用移液枪吸取1 mL上述缓冲液吹吸细胞沉淀,使其重悬,对悬浊液进行超声波破碎,整个反应过程始终处于低温状态进行。破碎后离心获得上清,上清液即为粗酶液,依据Shaw等 [16] 的方法对氯霉素酰基转移酶酶活进行测定。同时以BCA蛋白作为标准品,绘制标准蛋白曲线。最终计算氯霉素乙酰转移酶酶比活力。
2.6.3. qPCR反应
当菌液OD600为3时收集菌体沉淀,依照RNA提取试剂盒的操作步骤提取谷氨酸棒杆菌细菌总RNA,该试剂盒使用的方法为TRIZOL法。将提取出的RNA进行琼脂糖凝胶电泳以判定其是否完整,同时检测RNA浓度,以便后续试验时计算相关试剂的加入量。随后对提取的RNA进行后续操作,首先使用试剂盒去除谷氨酸棒杆菌基因组DNA,其次通过反转录形成cDNA。得到相应菌株的cDNA后使用绿色荧光染料以及qPCR检测系统Archimed-X4进行qPCR反应。本试验中每次反应阴性对照以及标准品校正的设置是为了确保试验结果有效,同时还需要进行三个生物学重复和三个技术重复,以进一步保障试验结果的有效性。根据文献记载,本试验中同样以谷氨酸棒杆菌的16S rDNA作为内参基因,ΔCT记作不同菌株CT值的差。氯霉素乙酰基转移酶作为报告基因,其表达水平表示的为带有重组启动子探测载体的谷氨酸棒杆菌中相应启动子的转录水平,16S rDNA基因的转录水平则表示野生型谷氨酸棒杆菌中启动子的转录水平 [17]。对数据进行处理是使用Livak等 [18] 的2−ΔΔCT方法,其中ΔΔCT是处理组,即构建的工程菌株的CT值的差减去对照组,即携带强启动子Ptac-M的谷氨酸棒杆菌的CT值的差,处理组ΔCT的计算方法为靶基因,即氯霉素乙酰转移酶的CT值与内参基因,即谷氨酸棒杆菌16S rDNA的CT值的差,对照组ΔCT值应用同样的方式处理数据。最终计算得出菌株C. glutamicum/pDXW-11-P1937和C. glutamicum/pDXW-11-Ptac-M的cat基因的转录水平。
3. 结果与分析
3.1. 二维电泳分离谷氨酸棒杆菌中蛋白
二维电泳技术的使用,使谷氨酸棒杆菌ATCC13032细胞中所含有的蛋白质得到分离,此步骤实验室前期工作中已经完成。二维电泳图如图1所示。详细内容参见张献 [13] 等文章。

Figure 1. The map of two-dimensional electrophoresis (the blots indicated by arrows with a MW size of approximately 30 KDa and a pI of approximately 4.0)
图1. 二维电泳图(箭头所指的MW大小约为30 KDa,pI约为4.0的斑点)
选择图1中箭头所指出来的蛋白点作为鉴定强启动子的点的原因为该斑点颜色较深,该蛋白斑点的MW约为30 KDa,pI约为4.0。实验室前期工作中已将上述标注出的蛋白斑点中所含有的蛋白质的分子质量以及等电点进行了鉴定,与所选的蛋白斑点匹配后,发现有6种蛋白质与二维电泳上的高表达蛋白斑点在理论上相匹配。其分别为dapF、gluB、aftC、Cgl0148、Cgl0392、Cgl1937、Cgl2540。其中dap蛋白家族、glu蛋白家族和aft蛋白家族含有单个蛋白,即dapF蛋白、gluB蛋白以及aftC蛋白,多重转录元件控制着它们的转录,所以难以预测准确的启动子序列。因此,Cgl1937、Cgl0148、Cgl0392以及Cgl2540基因被选择用来预测其相应启动子序列。
3.2. 网站预测启动子序列
利用预测网站对上述筛选出的编码相关蛋白质基因的启动子序列进行预测,发现未能预测出编码Cgl0148以及Cgl0392、Cgl2540基因的相应启动子序列。Cgl1937基因预测出了相应启动子序列,其序列为GTAACGTGGCAAAACGAACAATGTCTCACTAGACTAAAGTGAGATCGACA。如图2所示,ATG为Cgl1937基因的开放阅读框的起始密码子,TAA则作为终止密码子,序列总长度为72 bp。由此获得了Cgl1937基因的启动子P1937。
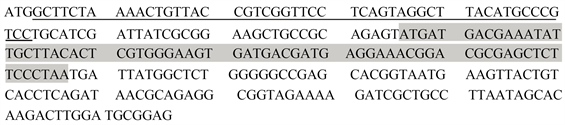
Figure 2. The ORF sequences of gene Cgl1937 (the grey part) and the sequences of the predict promoter (underlined nucleotides)
图2. Cgl1937基因开放阅读框(灰色部分)及预测的启动子序列(划线部分)
3.3. 工程菌株的构建
通过电击将扩增得到的启动子P1937和Ptac-M的DNA片段与启动子探测载体pDXW-11连接后的重组载体转化进谷氨酸棒杆菌ATCC13032感受态细胞中,抽提转化子DNA,对其进行验证时采用双酶切法。如图3所示,酶切后质粒大小达到了预期,说明成功构建并得到了携带启动子P1937的重组工程菌菌株C. glutamicum/pDXW-11-P1937和携带启动子Ptac-M的重组工程菌菌株C. glutamicum/pDXW-11-Ptac-M。
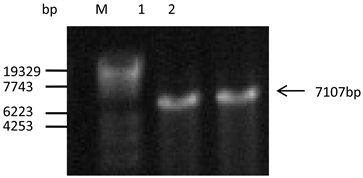 (M: Maker λ-EcoT14; 1: C. glutamicum/pDXW-11-Ptac-M; 2: C. glutamicum/pDXW-11-P1937)
(M: Maker λ-EcoT14; 1: C. glutamicum/pDXW-11-Ptac-M; 2: C. glutamicum/pDXW-11-P1937)
Figure 3. The result of double digestions
图3. 双酶切结果图
3.4. 鉴定内源启动子P1937转录活性
首先初步判定所含有的启动子的转录活性,使用氯霉素分别对野生型谷氨酸棒杆菌C. glutamicum ATCC13032以及在本研究中构建成功的工程菌菌株C. glutamicum/pDXW-11-P1937、C. glutamicum/pDXW-11-Ptac-M进行耐受性实验。实验结果表明,以上三种菌株的氯霉素耐受浓度分别为3 μg/mL、40 μg/mL以及30 μg/mL。随后为进一步研究两种启动子的转录活性,对工程菌株C. glutamicum/pDXW- 11-P1937和C. glutamicum/pDXW-11-Ptac-M的氯霉素乙酰基转移酶的比活力值进行检测,得到C. glutamicum/pDXW-11-P1937的CAT蛋白酶比活力为9.53 U/mg蛋白质;C. glutamicum/pDXW-11-Ptac-M的CAT蛋白酶比活力为0.85 U/mg蛋白质,结果见图4。
通过荧光定量PCR技术来直观的表现构建的工程菌菌株C. glutamicum/pDXW-11-P1937中所含有的启动子P1937和C. glutamicum/pDXW-11-Ptac-M中所含有的强启动子Ptac-M,对氯霉素乙酰基转移酶基因cat的转录水平,结果表明,启动子P1937控制下的cat基因的转录水平高于启动子Ptac-M控制下的水平,是其2.07倍,结果见图5。
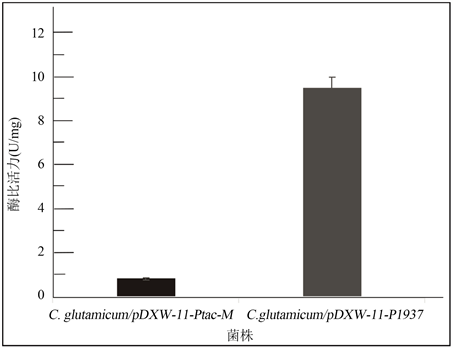
Figure 4. The detection of Chloramphenicol Acetyltransferase’s specific activity
图4. 氯霉素乙酰基转移酶酶比活力检测
4. 结论与讨论
本实验室的前期工作中,二维电泳技术的应用,使谷氨酸棒杆菌C. glutamicum ATCC13032的细胞中所含蛋白质得到了分离,选取其中一些高表达的蛋白点进行质谱解析,并通过在线启动子预测网站,对选出的蛋白点处的编码相应蛋白质的基因的启动子进行预测,相关文章详见张献等 [13]。前期构建的启动子Ptac-M为本试验所用的对照启动子,且其经过验证是谷氨酸棒杆菌强启动子 [12]。基于以上成果,为检测启动子转录活性的强弱,本研究设计了氯霉素耐受性实验及报告蛋白氯霉素酰基转移酶酶活实验。试验结果表明预测出的编码Cgl1937蛋白基因的启动子P1937的转录活性高于谷氨酸棒杆菌强启动子Ptac-M。而qPCR实验的结果则更加直观的表明在启动子P1937调控下cat基因的转录水平明显大于在启动子Ptac-M的调控下的转录水平。以上结果均可证明启动子P1937在谷氨酸棒杆菌中为强启动子。
为了更深入研究细菌的启动子,就需要人们识别和发现新的启动子,以满足人们的各种需求。本试验寻找谷氨酸棒杆菌内源启动子的方法为基于谷氨酸棒杆菌蛋白组的基础,通过启动子预测网站进行预测,以寻找更高效的内源启动子。此方法可以更加广泛的寻找启动子序列,为人们提供更多选择。除了通过上述方法寻找启动子,还可以基于启动子的结构特征、启动子在基因组中的分布特征、计算机建模、RNA聚合酶的特殊亚基等方式寻找新的启动子 [19]。
目前,对谷氨酸棒杆菌启动子的研究是一大热点,因其控制着转录的起始,且影响着谷氨酸棒杆菌蛋白表达系统能否高效表达目的蛋白。虽然目前关于谷氨酸棒杆菌内源强启动子的相关报道较少,但是会有越来越多的启动子元件被识别、被赋予功能,独特的启动子序列和有调控功能的元件可以被更合理的设计出来 [20]。本研究成功鉴定出高活性内源启动子P1937,为之后通过建立谷氨酸棒杆菌细胞工厂,利用其高效表达目标蛋白奠定了坚实的基础。
NOTES
*第一作者。
#通讯作者。