1. 引言
细胞凋亡在生物体的生长发育及新陈代谢中起着非常重要的作用,而凋亡过程的紊乱可能与许多疾病如肿瘤、自身免疫性疾病的发生有直接或间接的关系 [1] [2] [3] [4]。凋亡蛋白是指与细胞凋亡有关的蛋白质。研究表明,凋亡蛋白的功能与亚细胞位置密切相关 [5]。因此对凋亡蛋白亚细胞位置的正确定位,能帮助我们理解凋亡蛋白功能、细胞凋亡机制和药物开发。然而,通过传统的生物实验方法来确定凋亡蛋白的位置既费时又费力,难以满足现在的科研需求 [6]。因此,研究者开始借助计算机及其相关知识开发了许多有效且可靠的计算方法来替代或协助传统生物实验。
近年来,大量机器学习方法被开发用于识别不同的凋亡蛋白亚细胞位置,通常包括三个步骤:第一,从凋亡蛋白序列中提取包含不同种类蛋白质的信息作为凋亡蛋白亚细胞定位的特征向量,如信息增益(Increment of Diversity) [7]、位置特异性评分矩阵(Position Specific Scoring Matrix, PSSM) [8]、伪氨基酸组成(Pseudo Amino Acid Composition, PseAAC) [9]、氨基酸组成(Amino Acid Composition, AAC)和二肽组成(Dipeptide Composition) [10] [11]。近年来,随着特征提取方法的增多,以及计算机不断健壮的计算性能,许多研究者开始将多个特征进行融合以改进特征提取方法,如Zhang等人 [12] 通过将Moran自相关和互相关与PSSM集成在一起,提出了一种称为MACC-PSSM的新模型。又如刘等人 [13] 将氨基酸组成、二肽组成和自相关系数相结合来构建凋亡蛋白的特征表达模型。第二,将得到的特征向量输入到分类器中进行预测分类,在凋亡蛋白亚细胞定位中使用的分类器有协变判别函数法 [5]、模糊k-近邻 [14]、支持向量机(Support Vector Machin, SVM) [15] [16]、集成分类器 [17] 等。第三,通过Jackknife检验、K折交叉验证和独立集检验对分类器性能进行评估,以证明所提出方法的可靠性 [18] [19] [20]。这些计算方法的使用可以大大加快凋亡蛋白亚细胞位置的研究。这些方法都是基于序列提取得到的特征,好的特征提取方法对预测凋亡蛋白亚细胞位置是至关重要的,它能帮助我们提高预测准确率。
在本文中,为了能够更加准确的对凋亡蛋白亚细胞位置进行定位,我们考虑了凋亡蛋白序列的进化信息和序列信息。我们首先从序列中获取含有进化信息的PSSM,然后以一个分割比例将PSSM矩阵按行分割为两个子矩阵,并以此构建一个新的特征,我们称之为分割PSSM (Segmentation PSSM, SePSSM)。接下来我们对凋亡蛋白序列引入7种物化性质,并将此方法得到的特征与SePSSM特征进行线性融合。最后,我们将融合后的特征输入到含有四种不同核函数的支持向量机中,并通过Jackknife检验验证该方法的有效性。
2. 方法
2.1. 数据集
本研究使用了前人构建的两个基准数据集。由Zhou和Doctor [5] 构建的ZD98数据集包含43个细胞质蛋白(cytoplasm proteins, cy)、30个膜蛋白(membrane proteins, me)、13个线粒体蛋白(mitochondrial proteins, mi)和12个其他蛋白(other proteins, oth)。第二个数据集是由Zhang等人构建的ZW225 [15] 数据集,被分为4个亚细胞位置,包含70个细胞质蛋白(cytoplasm proteins, cy)、89个膜蛋白(membrane proteins, me)、25个线粒体蛋白(mitochondrial proteins, mi)和41个细胞核蛋白(nucleoid proteins, nu)。这两个数据集中的所有蛋白质序列均从SWISS-PROT中提取(http://www.ebi.ac.uk/swissprot/)得到 [21]。虽然两个数据集的数量较少,但在以往的研究中被广泛使用。
2.2. 特征提取方法
2.2.1. 从PSSM中获取分割PSSM(SePSSM)信息
首先我们使用POSSUM网页服务器(https://possum.erc.monash.edu/) [22] 生成PSSM,BLAST程序中参数选择为uniref50数据库,3次迭代,E-value 值为0.001,得到一个L × 20的矩阵。
(1)
其中,L表示蛋白质序列的长度,
表示蛋白质序列中氨基酸的进化信息。
接下来,为了从PSSM中获取更多重要信息,我们使用数学中的矩阵分块思想 [23],对PSSM矩阵进行按行分块,分割比例为
。因此一个L × 20的PSSM矩阵就被分割为
和
的两个子矩阵。由于只需将PSSM分割为两个子矩阵,因此对于分割比例
的选取为0到1之间的任意数(0和1都表示未分割),在实验中每次实验按比例增加0.1,因此
)。
最后,我们基于PsePSSM [24] 的部分思想,根据公式(2)分别计算被分割后的两个矩阵每列的平均值,以得到每一条凋亡蛋白序列对于20种氨基酸的平均突变概率。
(2)
其中,
为每个子矩阵的行数,即
。
最终我们得到一个40维的向量,我们将之称为分割PSSM(SePSSM)。
(3)
为了更好的理解SePSSM,我们以一条长度为1020的凋亡蛋白为例,则将得到一个1020 × 20的PSSM矩阵,下图为分割示意图如图1所示。根据这种对PSSM矩阵的分割和处理,我们将得到40维的SePSSM特征。
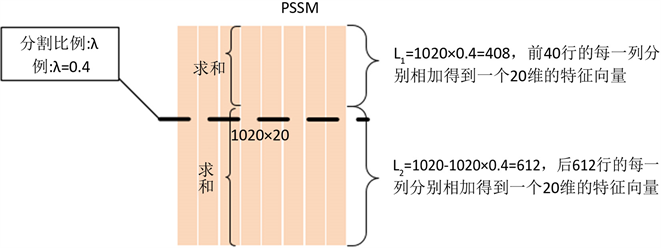
Figure 1. The segmentation diagram of SePSSM
图1. SePSSM分割图
2.2.2. 氨基酸的物理化学性质
氨基酸(AAs)的性质是由它们的侧链决定的,而这些侧链在形状、电荷和疏水性方面有所不同。因此,蛋白质可能具有不同的结构特征和生理功能。我们使用了原子吸收光谱的七种物理化学性质,包括归一化范德华体积、极性、溶剂可及性、变化、极化性、表面张力和二级结构 [25]。根据每种物理化学性质,20种氨基酸又被分为三组。这7种理化性质及其氨基酸的划分如表1所示。

Table 1. 7 kinds of physical and chemical properties and their division
表1. 7种物理化学性质及其划分
我们分别计算每一种物理化学性质的各类中所包含的氨基酸在蛋白质序列中出现的频率,因此每种物理化学性质将得到一个3维的特征向量。通过这七种物理化学性质,每一条凋亡蛋白序列我们将得到一个7 × 3 = 21维的特征向量。
2.3. 分类算法
提取特征后,可以使用各种分类算法来实现凋亡蛋白预测。在常用分类器中,由Vapnik提出的支持向量机表现出了很好的效果 [26]。支持向量机(Support Vector Machine)是一种按监督学习方式对数据进行二元分类的广义线性分类器,核心原理是找到一个分类超平面,以最大化正样本和负样本之间的距离,且利用核函数使它们在高维空间中线性可分离 [27]。SVM最初是为二分类设计的,而蛋白质亚细胞位置预测通常是个多类分类问题。但可以通过一对剩余(One vs Rest, OVR)和一对一(One vs One, OVO)等策略来实现SVM的多类分类,本文采用了前者OVR策略。由于选取不同的核函数,预测结果也会不同的。因此,本文将分别对四种常用核函数:径向基核函数(RBF)、线性核函数(linear)、sigmoid核函数(sigmoid)和和多项式核函数(poly)进行实验,以寻找最佳的核函数进行分类预测。
2.4. 评价方法
在统计学中,常见的三种检验方法为:自检验,Jackknife检验和独立集检验。Jackknife检验能得到唯一的预测结果,被认为是最客观、最严格的检验方法,且被广泛用于评价蛋白质亚细胞定位等领域中的预测性能 [28]。因此,在本文中,我们使用Jackknife检验方法和5种评价指标用于检验和评价我们所提出的预测凋亡蛋白亚细胞位置的预测模型方法。这5种评价方法是:敏感性(Sen)、F1值、马修相关系数(MCC)、准确率(ACC)和总体准确率(OA) [29]。公式如下:
(4)
(5)
(6)
(7)
(8)
表示属于第i类的样本被正确分到该类的数量,
表示属于第i类的样本没有正确分类到该类的数量,
表示被正确的分类到非i类的样本数,
表示非i类的样本错误的分类为i类的数量。
表示数据集中第i类的预测准确率。
表示对数据集中第i类的鲁棒性的度量,它是敏感性(
)和精度(
)的调和平均值,可以避免对某些指标的性能估计过高。
综合了不同的参数,能从整体上评价预测算法性能。
表示第i类在所有样本中预测正确的比率。
表示一个综合指标,它反映了总体的预测准确率,其中c为数据集中的类别数。以上5个指标的取值范围都在0到1之间,指标值越接近1,表示分类性能越好,指标值越接近0,表示分类器性能越差。
本文所提出的预测模型的流程图如图2所示,预测模型的详细描述如下:
· 获取序列。通过文献 [5] [15] 获得ZW225和ZD98两个数据集的氨基酸组成序列。
· 获得特征。通过POSSUM网页服务器获得两个数据集的PSSM矩阵,然后通过SePSSM方法(2.2.1节)从PSSM中获得40维的SePSSM特征,之后再与7种物化性质(2.2.2节)得到的21维特征进行融合,得到最终的61维特征。
· 预测分类。将得到的61维最终特征输入到含有不同核函数(RBF、poly、linear和sigmoid)的SVM中进行预测分类,并通过Jackknife检验得到最终的实验结果。
3. 结果和分析
3.1. SePSSM特征的分割比例
由于SePSSM特征是通过分割PSSM矩阵而得到的,而选择不同的分割比例
会得到不同的预测结果。为了得到最佳的结果,我们先对SePSSM特征进行分割比例的选择,每个分割比例在两个数据集上的总体准确率如下表2。为了证明分割的有效性,我们加入了不分割的实验结果,当
取1时,表示PSSM矩阵未进行分割。
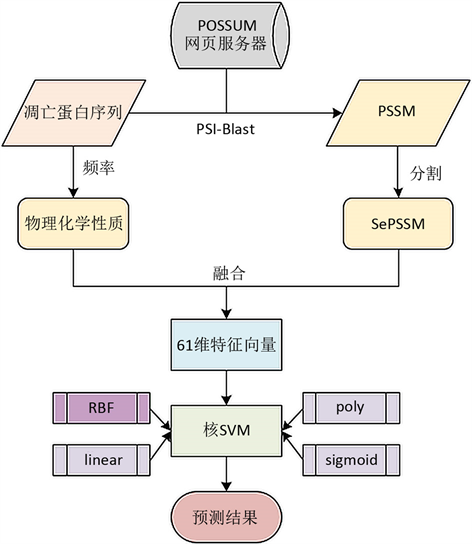
Figure 2. Flowchart of the proposed method
图2. 提出的方法的框架
Table 2. The OA(%) results of the two datasets at different segmentation ratios
表2. 两个数据集在不同分割比例的OA(%)结果
从表2我们可以看出,随着 的增大,ZW225数据集的OA值一直在波动,在
为0.5时SePSSM得到最好值81.7%;而ZD98数据集的结果先增大后减小,保持不变之后又减小,在
为0.5和0.6时SePSSM结果最好,最好值为92.9%。且从结果中可以看出,对PSSM矩阵进行适当的分割要比未分割的结果好,这是因为分割之后将数据细化,获得了更多有用的特征信息,而这些信息更能对每个类进行区分,从而有利于进行预测分类。当分割比例为0.5时在ZW225和ZD98两个数据集上得到的结果要比未分割或其他分割比例的结果好。因此,本文中的分割比例取0.5。
3.2. 不同特征的性能
为了验证我们所提出的SePSSM的性能,我们计算了SePSSM、PsePSSM [24]、7种理化性质(PhyChe)和融合特征分别在ZW225和ZD98两个数据集上的总体准确率,所有结果在同一条件下通过Jackknife检验得到,结果如下表3。
Table 3. The OA(%) of different feature methods on two datasets
表3. 不同特征方法在两个数据集上的总体准确率(%)
由于PsePSSM中含有一个决定特征维度的参数,在序列较长时该参数的选择将会耗费较多的计算时间。而从表3可以看出,SePSSM在PSSM上进行分割就能达到一个较好的预测效果,且在ZW225数据集上比PsePSSM高7.6%。这表明我们提出的SePSSM的性能要优于PsePSSM,且计算方法省去了PsePSSM计算其他特征的时间,这将更加简便。将SePSSM和7种理化性质融合的预测结果要优于单一的特征,达到了一个较好的预测值,分别为94.6%和96.9%。
3.3. 不同核函数对结果的影响
在使用支持向量机进行分类的时候,可以对SVM的核函数进行选择,选取不同的核函数会对结果产生较大影响。因此本文在融合SePSSM和PhyChe对凋亡蛋白序列进行特征提取,SePSSM的分割比例为0.5。选取不同的核函数在两个数据集上的准确率如下表4和表5。
Table 4. The prediction results of ZW225with different kernel functions
表4. ZW225在不同核函数下的预测结果
Table 5. The prediction results of ZD98 with different kernel functions
表5. ZD98在不同核函数下的预测结果
从表4和表5可以看出,对ZW225和ZD98两个数据集,用SVM进行分类时,采用RBF核函数和linear核函数的预测效果要优于poly和sigmoid两种核函数,且采用RBF核函数预测总体准确率要略高于linear核函数,sigmoid核函数得到的效果最差。因此,本文选用RBF核函数作为SVM算法的核函数。
3.4. 我们提出的方法性能
本文通过融合PsePSSM和PhyChe两种特征,SVM的核函数选择RBF,通过Jackknife检验对ZW225和ZD98数据集进行验证,得到的结果如表6所示。从表6可以看出,我们的方法对ZW225和ZD98数据集的OA分别达到了94.6%和96.9%。实验结果表明,该方法能够有效预测凋亡蛋白的亚细胞位置。
Table 6. The predictive performance of datasets ZW225 and ZD98 protein subcellular localization on the Jackknife test
表6. ZW225和ZD98数据集的蛋白质亚细胞定位在Jackknife检验下的预测性能
3.5. 与其他方法的比较
为了进一步评估我们方法的有效性,我们将我们提出的方法与几种凋亡蛋白亚细胞定位的方法进行了比较。表7和表8分别显示了不同方法对ZW225和ZD98两个数据集的OA和每类亚细胞位置的敏感性的预测结果,所有结果都是通过Jackknife检验得到的。
Table 7. Comparison from different methods on ZW225 dataset by Jackknife test
表7. ZW225数据集基于不同方法的预测结果
Table 8. Comparison from different methods on ZD98 dataset by Jackknife test
表8. ZD98数据集基于不同方法的预测结果
从表7和表8可以看出,我们提出的方法在两个数据集的cy类上都取得了较好的结果,分别为98.6%和97.7%,这是因为这两个数据集类中数量最多的都是cy类,从而导致了预测效果较好。而nu和oth类,分别在两个数据集中数量都最少,使得训练不足导致预测性能较差。但从OA值来看,我们的方法要高于其他方法的,这表明我们的方法具有较好的预测性能。
4. 结论
首先,基于矩阵分块的思想从PSSM中提取SePSSM特征,然后将SePSSM和7种理化性质得到的特征融合构建凋亡蛋白序列的特征表示方法,通过实验结果可知,对PSSM进行平均分割比不分割或其他的分割比例的预测效果更好。最后,ZW225和ZD98两个数据集在RBF核的SVM分类器上分别进行预测分类,分别得到了94.6%和96.9%的总体准确率,这已高于大多数已有的凋亡蛋白亚细胞定位算法,这表明我们所提出的方法是可行的。鉴于我们使用的数据集为不平衡数据集,数据集类中数量存在较大差异,因此在下一步研究中,我们将考虑对数据集进行采样处理或构建一个平衡的数据集来对凋亡蛋白进行预测研究。
基金项目
国家自然科学基金(62062067)。