1. 引言
最近,研究人员发现人类语言与生物语言之间存在一些相似之处。受此启发,开发生物语言处理模型可以为生物序列的研究提供一个全新的理论视角和可行的方法,然而,人们要发现与人类语言不同的生物模式的真正含义是相当困难的。研究人员开发了一个叫的BioVec [1] 模型。该模型提供了一种全新的蛋白质序列的表示和特征提取方法。Wang等人开发了一种名为生物向量(Bio2Vec)的生物语言处理模型 [2]。Bio2Vec提供了一个框架,允许研究人员考虑生物序列的上下文信息和隐含语义信息。Chen等人 [2] 利用自然语言处理(NLP)技术获得了各蛋白质序列的全局向量表示。Heinzinger等 [3] 利用深度双向语言模型ELMo [4] 从未标记的大数据中捕捉蛋白质序列的生物物理特性。MENEGAUX等人 [5] 开发了一称之为fastDNA的模型,此模型通过学习DNA序列所包含的k-mer的连续低维表示,将DNA序列嵌入到一个向量空间中。他们通过修改fastText开源库 [6] [7] 实现了这个模型,其中涉及到一个类似的自然语言的k-mer嵌入模型。Patrick Ng开发了一个名为DNAVec [8] 的方法,该方法基于NLP的一个开创性的模型Word2Vec [9] 来提取DNA序列的特征并给出适当的表示,可以更好地理解DNA序列的语义。以上实验结果表明,利用生物序列的语义信息对解决基于序列的问题有很大帮助。此外,这些工作将在各种生物分类问题上也有潜在的应用。
上面研究的表明NLP在生物语言处理领域有很大的潜力,但是自然语言处理社区技术也不断的更新和发展,以前技术的不足也不能满足当下发展的需求。比如:之前的模型Word2Vec [9]、GloVe [10] 专注于学习与语境无关的词语表征,最近的研究ELMo [4] 使用了一个双向的语言模型学习上下文表示,集中在学习依赖语境的单词表征上,ELMo使用两个独立的长短期记忆网络(LSTM) [11] 的组合,并不是真正意义上的上下文相关表示。GloVe [10] 使用机器翻译将上下文信息嵌入到单词中。这些自然语言处理技术虽然得到了广泛的应用,但它们对序列的表示都不是真正意义上的上下文相关。BERT [12] 是广泛用于NLP领域的基于上下文相关的词表示模型,同时在大多数的NLP应用中取得了最先进的性能。在大量自然语言处理领域的实践表明BERT具有很强的表示能力,而表示学习 [13] 对于深度学习模型是非常重要的。基于BERT这种对序列的表示能力,因此在这篇文章中我们基于全基因组DNA序列从头训练BERT模型,从而得到一个DNA序列的表示模型。模型的可视化结果表明,模型能够捕捉到DNA序列中特殊的语义模式。
2. 预训练模型
2.1. 数据集
我们的预训练模型是用hg38人类基因组组装chr1至chr22进行训练的。具体来说,它们是从UCSC (http://hgdownload.cse.ucsc.edu/downloads.html#human)下载的。此外,我们排除了X和Y染色体,以及线粒体和未定位的序列。
2.2. BERT:基于Transformer的双向语言模型
BERT是一个上下文相关的的词表示模型。该模型是基于遮蔽语言模型和预训练的使用双向Tranformers [14]。由于语言模型的本质是未来的词不能被看见,以前的语言模型仅限于两个单向语言模型的组合(即从左到右和从右到左)。BERT使用了一个遮蔽语言模型,可以预测序列中随机遮蔽的词,因此可以用于学习上下文相关表示。同时,它在大多数NLP任务上获得了最先进的性能,同时只需要最小的特定任务架构修改就能用于其他任务。根据BERT的作者,在自然语言模型中加入双向表征的信息,而不是单向表征的信息,对于表征自然语言中的词是至关重要的。我们假设这种双向表示对于DNA序列的表示也是至关重要的。因为复杂的DNA序列表示也不是简单的从左到右或者从右到左的关系,基因调控元件之间也是上下文相关的。
2.3. 分词
我们没有将每个碱基视为一个单一的标记,而是用k-mer表示法将一个DNA序列标记化,这种方法已被广泛用于分析DNA序列。k-mer表示法通过将每个脱氧核苷酸碱基与它的后续碱基连接起来,为其整合了更丰富的上下文信息。它们的连接称为k-mer。本文中我们使用可变长k-mer (
)来组装DNA序列。具体方法如下:给定一个DNA序列S,首先通过在S上滑动长度为k的窗口将其转换为重叠的固定长度的k-mer,其中k值的选择采用离散随机采样。例如,GATCCCAC的变长k-mer (k = (4, 5, 6)可以是{GATC, ATCCC,TCCCAC}。分词示例如图1所示。在我们的实验中,模型的词汇表包括k-mer的所有排列组合以及5个特殊标记。[CLS]代表分类标记;[PAD]代表填充标记;[UNK]代表未知标记;[SEP]代表序列上下句分离标记;[MASK]代表屏蔽标记。因此模型中的词汇共有87365个。
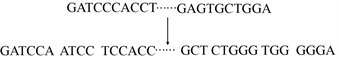
Figure 1. Variable length k-mer assembled
图1. 可变长k-mer从头组装
2.4. 预训练
根据之前的模型预训练工作 [12] [15],本实验的总体结构如图2所示,对于一段DNA序列(序列的最大输入长度为512),我们将其标记为k-mers序列,并在其开头添加一个代表整个序列的特殊的标记[CLS]以及在结尾添加一个表示序列结束的特殊的标记[SEP]。在训练过程中,我们遮蔽序列中的某些k-mers,遮蔽的比例为占一段输入序列的15% (防止过拟合以及减少模型的计算量)。在本研究中我们使用BERT (L = 12, H = 512, A = 12)相同的模型结构进行训练,其中L代表代表模型的总的层数,即有12个Transformer结构单元,H代表隐藏层大小,A代表自注意力头部,共有12个注意力头部。我们对预训练模型进行了共80 k步的训练,批处理量为8。学习率为4e−4。此外,我们在配备2个NVIDIA Tesla K80 (240 k) GPU的机器上进行训练。
2.5. 词频–逆文档频率
tf-idf (term frequency-inverse document frequency)表示词频–逆文档频率,tf (term frequency)是词频,idf (inverse document frequency)表示逆文档频率,tf-idf权重是信息检索和文本挖掘中常用的加权技术。该权重是一种统计度量方法,用于评估一个单词对集合或语料库中的文档的重要性。重要性随单词在文档中出现的次数成比例增加,但会被单词在包含该单词的语料库中出现的频率抵消,这有助于根据某些单词在一般情况下出现更频繁的事实进行调整。tf-idf计算方法为:
我们定义
为单词t在文档d中的出现频率,然后词频
如公式2-1所示:
(2-1)
逆文档频率idf定义如公式2-2所示:
(2-2)
其中n 在文档集中文档的总数,
是包含t的文档的数量,
定义如公式2-3所示:
(2-3)
然后将得到的tf-idf向量用欧几里得范数进行归一化处理,如公式2-4所示:
(2-4)
3. 实验结果
3.1. 模型注意力机制分析
BERT以两个关键思想为基础:Transformer [14] 架构和无监督的预训练。这两个思想是BERT在很多应用上取的很好的原因。Transformer是一个序列模型,它放弃了循环神经网络(RNN) [16] 的循环结构,而采用了完全基于注意力的方法 [17]。为了探索预训练到底学到了什么。我们使用可视化工具bertviz [18] 对模型进行可视化,通过分析模型的注意力机制我们发现一些非常独特的和令人惊讶的直观的注意力模式。下面我们发现了4种比较重要的注意力机制模式,并为每一个特定的layer/head进行分析。
模式一:注意力大部分放在3-mer上。在这个模式中,在特定位置的大部分注意力指向序列中的3-mer标记。我们可以在layer 0/head 3中看到这样的一个例子。(选中的头部由顶部颜色条中突出显示的正方形表示),图3(a)显示了对一个选定标记mer“gatccac”的注意力模式。在本例中,几乎所有的注意力都指向序列中的下一个令牌“cct”、“aaa”、“cct”。一个词注意放在3-mer,可能是因为3-mer是密码子的,在一个序列中比较重要的序列单元的原因。

Figure 3. Visualisation of attentional mechanisms in pre-trained models
图3. 预训练模型中的注意力机制可视化
模式二:在这个模式中,序列是一段管家基因序列。注意力放在了除3-mer之外的所有标记。例如,在图3(b)中layer3,head3中,“gcgc”注意放在任何不包含“gat”、“aaa”、“cct”的k-mer。一项研究用k-mer频率作为特征来预测CpG岛,结果表明人类的CpG岛序列具有独特的k-mer模式,并不是随机序列,通过他们的分析,4-mer的表现最好 [19]。表明CpG岛具有明显的4-mer分布。正如我们所知道的,CpG岛富集于管家基因,管家基因因为要维持在所有细胞中稳定表达,而3-mer是比较常见的转录密码子,在基因组属于转录比较活跃的部分。因此,模式二可能学习到序列中隐含的调控语义。
模式三:注意力放在了语料库中tf-idf值比较高的k-mer上。我们计算了全基因组DNA序列语料库中的k-mer的tf-idf值。其中tf-idf值前100的k-mer的词云图如图4所示。根据图3(c)可知,注意力机制放在了tf-idf值比较高的mers上。模式三表明该模型能捕捉到语料库中的关键k-mer。
模式四:在这一模式中。注意放在了相同或相关的词,包括源词本身。在图3(d)的例子中,“gagtgagt”的注意力主要集中在它本身和“gtgctg”、“tgctgga”上。这种模式不像其他模式那样明显,注意力分散在许多不同的单词上,但是其注意力类似于自然语言领域把注意力放在近义词上,表明模型能够捕获相似的语义。以上4种模式描述了预训练模型中比较常见的注意模式,结果表明该模型实际上得到了一些有意义的DNA序列特性。

Figure 4. Word cloud graph of the top 100 kmer with relatively high tf-idf values
图4. 前一百个tf-idf值比较高的kmer的词云图
3.2. DNA序列的词向量表示
长DNA序列的普遍表示之一是将其分解为较短的k-mer成分,在许多应用中,短k-mer被认为是相关的,如k = 6 [2],k ≤ 7 [20],k = 8 [21]。不幸的是,将k-mer作为一个one-hot向量进行直接的编码时任何一对one-hot之间的距离都是等距的,这表示k-mer之间完全没有相关性。而且这种编码常常导致数据的空间维度很大,容易受到维数诅咒的影响。当应用最新的机器学习算法来解决生物序列分析的问题时,这尤其成问题。因此,我们提出了一种基于预训练的DNA序列表示方法。考虑到BERT用于预训练的字典不能太大,我们使用长度为3~8的k-mer。训练模型将每个k-mer嵌入一个新的n维特征空间,为不同长度的k-mer生成特征向量。具体地,预训练嵌入模型可以表示为大小为
的投影矩阵,其中V为词典大小,N为嵌入特征空间的维数,词汇表V是所有k-mer的组合和用于标记的5个特殊符号的集合。在本论文中,我们提取预训练模型的最后一层隐含层,得到一个大小为
的嵌入矩阵。每个单词都嵌入在一个512维的空间中。我们可以使用一个名为bert-as-service的工具(https://github.com/hanxiao/bert-as-service)获取上下文单词嵌入。获得的预训练的词向量形式如图5所示。

Figure 5. Pre-trained word vector representation
图5. 预训练词向量表示形式
4. 结论
目前预训练技术广泛应用于自然语言处理(NLP)和计算机视觉(CV)领域。但是在生物信息领域还很少应用到。DAN序列作为生物语言,其本身和自然语言有一定的相似性。因此本论文试图从语言处理的角度分析生物语言。希望为DNA序列的解读提供新的发现或为生物问题的解决提供新的视角。在本研究中,通过自监督的预训练方法来得到DNA序列的上下文相关表示,这与之前的序列one-hot方法表示或者序列上下文无关表示极为不同。论文的主要贡献是发布了一个预训练模型,我们期望我们的模型也适用于其他序列分析任务,例如,从染色质可及性测序数据 [22] 和转录因子结合位点测序数据 [23] 中确定基因组调控元件。此外,由于RNA序列与DNA序列只相差一个碱基,而语法和语义基本保持一致,我们提出的方法预计也可能应用RNA序列数据 [24]。虽然在DNA上像自然语言样直接进行机器翻译是不可能的,但DNA序列的预训练模型提供了这种可能性的启示。作为一个基于DNA序列开发的预训练语言模型,它正确地捕捉到了DNA序列中隐藏的语法和语义。同时,本论文也凸显了结合不同层次的数据对DNA序列进行解读的必要性。综上所述,我们预计此预训练模型可以为基因序列分析带来先进的语言建模视角,为生物信息学界带来新的见解。
参考文献