1. 引言
川崎病(Kawasaki Disease, KD),又称皮肤黏膜淋巴结综合征,是一种以全身血管炎为主要病变的急性发热出疹性疾病,其特征为广泛的中小血管炎症,以心血管系统的损害最为严重。川崎病又分为典型性川崎病(Typical Kawasaki Disease, TKD)和不完全川崎病(Incomplete Kawasaki Disease, IKD)。噬血细胞综合征(Hemophagocytic Syndrome, HPS)又称嗜血细胞淋巴组织细胞增生症或嗜血细胞性网状细胞增生症(HLH),也是一类由免疫紊乱介导的疾病,该病分原发性和继发性。川崎病合并噬血细胞综合征近年来偶有报道,本文回顾性分析1例免疫球蛋白无反应型不完全性川崎病并嗜血细胞综合征患儿临床资料和相关文献,以希望提高对该病的认识及早期识别,从而达到早期治疗及提高预后。
2. 临床资料
患儿,男,2岁11月,因“发热伴轻咳半个月,皮疹1周”入院。患儿入院前半个月无明显诱因出现发热,中高热为主,每天4~6次,口服退热药难以降至正常,伴轻咳。入院前1周出现全身散在红色皮疹,伴瘙痒,唇略红,先后予哌拉西林舒巴坦、阿奇霉素抗感染治疗,外院也曾考虑不完全川崎病,在发病第10天使用免疫球蛋白30 g (2 g/kg)治疗,但效果欠佳,仍反复发热,遂收入我院进一步诊治。入院体查:T:38.6℃;P:140次/分;R:56次/分;BP:96/56 mm Hg;体重:15 kg;身高:96 cm;神清,精神可,全身躯干散在皮疹,部分结痂,颈部红色皮疹,颈部触及数枚肿大淋巴结,最大1.0 × 2.0 cm,活动度可,边缘尚规整,球结膜无充血,唇略红,草莓舌,双肺呼吸音粗,未闻及干湿啰音,心音有力,律齐,未闻及杂音。腹软,肝脾未触及肿大,神经系统检查未见异常,双下肢略水肿,四肢末端无硬肿及脱皮,肛周无脱皮。
该患儿入院时我科仍然考虑不完全性川崎病,入院第2天(发病第16天)继续免疫球蛋白2 g/kg冲击治疗,鉴于外院曾使用过免疫球蛋白冲击治疗后仍发热,不排除同时合并感染,继续使用了舒普深治疗后联合万古霉素治疗,但患儿仍反复高热。
实验室检查:CRP 123.8 mg/L (<8.21 mg/L),血常规:WBC 24.4 × 109/L (5~12 × 109/L),N 76% (40%~60%),HB 101 g/L (105~145g/L),PLT 403 × 10/L (140~440 × 10/L),中性粒细胞杆状核8% (0%~2%)。尿液分析及粪便常规 + 隐血未见异常。白蛋白30.5 g/L (40~55 g/L),hs-CRP 136.27 mg/L (0~6 mg/L)。ESR 41.0 mm/H (0~15 mm/H)。PCT 0.641 ng/ml (<0.1 ng/nl)。免疫6项示IgG 22.00 g/L (5.0~10.6 g/L),IgA 1.41 g/L (0.34~1.38 g/L),IgM 1.46 g/L (0.44~1.44 g/L),C3 1.88 g/L (0.8~1.5 g/L),IgE 319 IU/ML (0~100 IU/ml)。FER 14,242.00 ng/mL (10~291 ng/ml)。凝血四项、肝酶、心肌酶、肾功能、淋巴细胞计数正常。病原学:病毒血清学试验示:EBVCA-IgM阴性,EBVCA-IgG阳性,EBVEA-IgG阴性,EBVNA-IgG阳性,EBVIgG抗体低亲和力弱阳性、高亲和力阴性,血浆EBV-DNA < 500 cps/ml (检测下限500 cps/ml);九项呼吸道病原体抗体IgM均阴性,咽拭子I + II类阴性;骨髓培养、脑脊液培养、肥达、外斐氏反应、ASO、尿及粪便培养、真菌二项、结核T-spot等检查均阴性。自身抗体18项、血管炎4项、自免3项、肿瘤标志物均阴性。
影像学检查:胸片提示支气管炎。入院第1天腹部超声示肝、胆、脾、胰、双肾未见明显异常,消化道积气,腹腔少量积液。入院第10天腹部超声示肝脾大,胆胰未见异常。入院当天(病程第15天)心脏彩超示左、右冠状动脉内膜稍增粗(图1)。入院第11天(病程第26天)心脏超声提示:左右冠状动脉管周组织灰度增强,内膜尚光滑,心包积液(微量) (图2)。全腹部MRI + 头颅MRI检查排除肿瘤及占位性疾病。
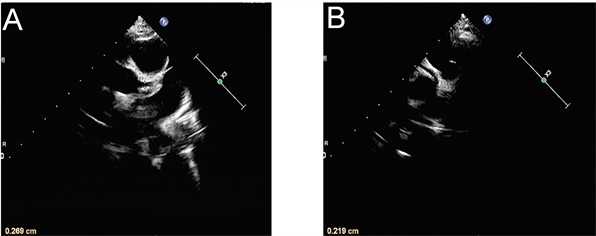
Figure 1. Cardiac ultrasonography on day 15 of the course of disease: The diameter of coronary artery was 2.7 mm (z = 1.05) for the left coronary artery (A) and 2.2 mm (z = 0.59) for the right coronary artery (B).
图1. 病程第15天心脏超声:冠状动脉内径分别为A:左冠状动脉2.7 mm (z值1.05)和B:右冠状动脉2.2 mm (z值0.59) (病程第15天心脏超声结果)。
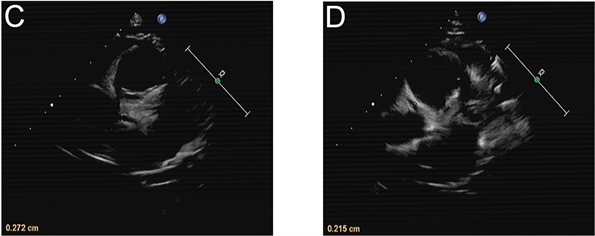
Figure 2. Cardiac ultrasonography on day 26 of the course of disease: The diameter of coronary artery was 2.7 mm (z = 1.05) for the left coronary artery (C) and 2.2 mm (z = 0.59) for the right coronary artery (D).
图2. 病程第26天心脏超声:冠状动脉内径分别为C:左冠状动脉2.7 mm (z值1.05)和D:右冠状动脉2.2 mm (z值0.59) (病程第26天心脏超声结果)。
入院第7天体查发现肝脾逐渐增大,复查血常规提示WBC 6.7 × 109/L,HB 82 g/L,PLT 55 × 109/L,三系出现明显下降,考虑有嗜血细胞综合征倾向,病情进展,完善嗜血细胞综合征相关实验室指标。检查提示:FIB 0.6 g/L (2~4 g/L),ALT 365 IU/L (9~50 U/L),LDH 7161 IU/L (159~322 U/L),FER > 16,500 ng/mL (10~291 ng/ml)。甘油三脂1.80 mmol/L (0.23~1.70 mmol/L),淋巴细胞计数明显下降,NK细胞活性减低,sCD25 19,343 pg/ml (<6400 pg/ml)明显升高;骨髓检查提示增生低下,组织细胞比例明显增高,吞噬现象易见;HLH突变基因检测:该患者未检索到与HLH相关变异基因位点,主要检测以下基因位点:PRF1、UNC13D、STX11、STXBP2、XIAP、SH2D1A、RAB27A、MY05A、AP3B1、LYST、ITK、SLC7A7、BLOC1S6、CD27、MAGT1、ADA、BTK、IL2RA、IL2RG、MVK、PNP、WAS、FAS、RECQL4、RAG1、RAG2 (这些是明确的HLA相关基因的纯合或复合杂合突变,或行WES发现其免疫调节基因的缺陷)。颈部淋巴结活检提示淋巴结组织细胞增生性病变,以组织细胞增生性淋巴结炎改变为主(图3)。
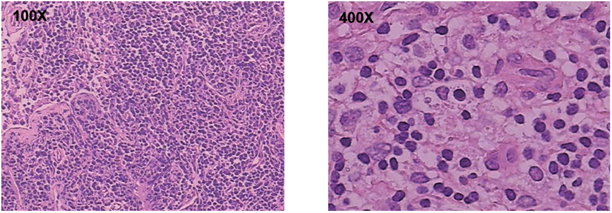
Figure 3. Neck lymph node biopsy results: Microscopically, the lymph nodes of the left neck were not clear, mainly mature tissue cells and proliferation of T cells, no obvious necrosis was observed, and a small amount of vacuolation of tissue cytoplasm was observed. Immunohistochemistry: CD3+, Bcl-2 part +, CDA−, Langerin−, MPO scattered in small amount +, Ki67 about 30%+, CD10 part +, CD79A−, CD68+. Pathological diagnosis: histiocytic proliferative lesions in the left cervical lymph nodes, mainly histiocytic proliferative lymphadenitis.
图3. 左颈部淋巴结镜下见淋巴结结果欠清,以成熟的组织细胞及T细胞增生为主,未见明显坏死,可见少量组织细胞质空泡化。免疫组化:CD3+、Bcl-2部分+,CDA−,Langerin−, MPO散在少量+,ki67约30%+,CD10部分+,CD79a−,CD68+。(颈部淋巴结病理活检结果)
病理诊断:左颈部淋巴结组织细胞增生性病变,以组织细胞增生性淋巴结炎改变为主。
根据2004年国际组织细胞学会修订的HLH诊断标准 [1]:1) 发热超过1周,热峰 > 38.5℃;2) 脾大;3) 两系或三系血细胞减少(血红蛋白 < 90 g/L,血小板 < 100 × 109/L,中性粒细胞绝对值< 100 × 109/L)且非骨髓造血功能减低所致;4) 血三酰甘油升高(≥3 mmol/L)或高于同年龄的3个标准差和(或)纤维蛋白原下降(<1.5 g/L)或低于同年龄的3个标准差;5) 血清铁蛋白升高(≥500 μg/L);6) 血浆可溶性CD25 (可溶性IL-2受体)升高(≥2400 IU/ml);7) NK细胞活性下降或缺乏;8) 骨髓、脾脏、脑脊液或淋巴结发现噬血细胞现象,未见恶性肿瘤细胞。而根据Hscore评分系统 [2],该患儿发生HLH概率99.8%。该患儿嗜血细胞综合征诊断明确,考虑不完全性川崎病合并嗜血细胞综合征,按HLH-2004方案给予化疗,2周后患儿临床症状消失,查体肝脾未触及肿大,复查实验室指标明显好转,于入院后43天出院,目前定期在我院血液科化疗,随访3个月,未见复发。
3. 讨论
完全性川崎病( TKD)已有明确的诊断标准 [3] [4],但关于不完全性川崎病(IKD),目前尚没有明确的诊断标准,根据美国心脏协会的诊断建议 [4],满足以下条件者可诊断:具有发热 ≥ 5 d,仅有2项或3项临床特征,除外渗出性结膜炎、渗出性咽炎、溃疡性口腔炎、大疱性或水疱性皮疹、全身淋巴结肿大或脾肿大;婴儿发热 ≥ 7 d且无其他原因可以解释者,需要考虑不完全KD的可能。同时AHA也提出了不完全川崎病的诊断流程,在儿童发热 ≥ 5 d并且满足2条或3条诊断标准或婴儿发热 ≥ 7 d无其他原因可以解释时,要进行实验室评估,如伴CRP > 30 mg/L和(或) ESR > 40 mm/H,并满足6项附加的实验室检查标准中的3项则可以诊断不完全性川崎病 (主要包括:血清白蛋白 ≤ 30.0 g/dl;贫血;谷丙转氨酶升高;病程7 d 后血小板水平 > 450 × 109/L;白细胞 > 15 × 109/L;尿白细胞 > 10个/HP),或者不满足该6项中的3项,而伴有冠状动脉改变者 [4] [5]。噬血细胞综合征(hemophagocytic lymphohistiocytosis HLH)是一类由免疫紊乱介导的疾病,其发病机制目前尚未完全阐明,临床上分为原发性和继发性两大类。原发性HLH是一种常染色体或性染色体隐性遗传病。目前报道的明确与HLH相关的基因有12种,根据缺陷基因的特点将原发性HLH分为家族性HLH (FHL)、免疫缺陷综合征相关HLH和EB病毒(EBV)驱动HLH [6]。而继发性HLH则与各种潜在疾病有关,是由感染、肿瘤、风湿性疾病等多种病因启动免疫系统的活化机制所引起的一种反应性疾病,通常无家族病史或已知的遗传基因缺陷。
本次报道的该病例,根据AHA诊断标准,入院时发热超过5天,同时合并有唇红、皮疹及淋巴结肿大,实验室指标CRP > 30 mg/L、ESR > 40 mm/H,伴贫血、血小板水平 > 450 × 109/L,白细胞 > 15 × 109/L,为此符合不完全型川崎病的诊断。外院第一次IVIG治疗无反应,但实际上IVIG治疗无反应患者于初次注射完IVIG后仍持续发热36 h或以上,或者再度发热,2017-03-29美国心脏协会于网上发布了2017年版《川崎病的诊断、治疗及远期管理——美国心脏学会对医疗专业人员的科学声明》 [4] 建议应用第二剂IVIG (2 g/kg) (IIa类,B级),或大剂量甲泼尼松龙冲击治疗(IIb类,B级)等治疗措施。但该患儿先后两剂IVIG冲击治疗均无效,同时由于家属的不配合也未能早期激素治疗。该例患儿疾病初期各实验室检查并未提示HLH,随着疾病进展,患儿出现反复高热不退,肝脾进行性增大,伴转氨酶明显升高,外周血3系减少,低纤维蛋白原血症,淋巴细胞计数提示T细胞、NK细胞等绝对计数明显下降,NK细胞活性下降,血清铁蛋白明显增高,血浆可溶性CD25升高,骨髓涂片检查见吞噬细胞,按照HLH-2004诊断标准 [2],后期完全符合嗜血细胞综合征的诊断。
HLH作为KD的一种并发症有文献又称为巨噬细胞活化综合征(Macrophage activation syndrome MAS),也研究者被称为继发性吞噬性淋巴组织细胞增多症(HLH)。HLH作为川崎病的严重并发症,两者早期有许多重叠之处 [7]。目前有文献报道认为是免疫系统由于感染激活引起 [8]。对于继发性HLH早期诊断及治疗对于阻断其严重的致命性后果有重要影响。Jung Eun Choi等回顾分析247例不完全川崎病临床资料,其中有8例发生HLH,发现出现HLH的8例患者中,其发热时间明显延长,血清铁蛋白(发生HLH组均值3122 ng/ml,而未发生HLH组均值120.1 ng/ml)及N-脑钠肽(NT-poBNP)明显升高 [9]。FER是一种急性期反应物,在炎症或感染性疾病中经常升高。此外,如果FER水平异常显著升高,常与特定的自身免疫性疾病,以及某些神经疾病和恶性肿瘤有关。Nasir等报道的3例不完全性川崎病患儿的FER会显著升高(7065 pmol/L、33,373 pmol/L、17,304 pmol/L) [10]。为此有学者认为FER水平的显著增加,尤其是当它大于5000~10,000 ng/ml时,它是MAS的一个重要诊断生物标志物 [11]。而在疾病评估治疗反应和预测预后方面,对单个患者FER水平的连续监测其意义更大 [12]。Kostik等人认为可以把以下3个以上的实验室变量进行组合是早期诊断的可靠指标:如血小板和白细胞计数减少;白蛋白和纤维蛋白原水平降低;铁蛋白、天冬氨酸氨基转移酶和乳酸脱氢酶水平升高;以及存在蛋白尿的情况 [13]。回顾上述病历,该患儿入院时已经发热半月余,表现为反复高热,入院时诊断不完全性川崎病,查FER显著升高,转氨酶正常高值,乳酸脱氢酶偏高,其他HLH指标并不明显,我们再次利用Hscore评分系统 [2] 进行评分提示该患儿入院时发生嗜血的概率大约是4.4%,提示该患儿随着病情的进展,发生嗜血概率较大,应该早期干预及治疗。
KD为多系统损害的发热性疾病,合并MAS的临床表现往往与原发病活动难以区分。有研究者认为KD合并MAS与自身免疫性疾病如s-JIA合并MAS有相似病理生理基础,自身免疫性疾病或传染源的高活性触发,导致细胞毒性T细胞和巨噬细胞活化后免疫激活时间延长,引起全身炎症因子风暴,这些细胞因子包括干扰素γ (IFNγ)、肿瘤坏死因子ɑ (TNFɑ)、IL-2、IL-1、IL-6、IL-18以及巨噬细胞集落刺激因子(M-CSF) [14] [15] [16]。但实际上,KD中的MAS发生率低于SJIA和SLE,估计约为1.1% [17],而Latio及其同事报道KD合并MAS发生率为1.9% [18]。但郭莉等研究者发现,相对于全身型幼年特发性关节炎(SJIA)以及结缔组织病(CTD),KD合并巨噬细胞活化综合征(MAS)发生时间却最快,KD合并MAS多于起病5天左右,提示KD起病早期即可能合并MAS,甚至可直接以MAS起病 [19]。而国外研究者发现KD合并MAS发病时间约为13.3天(3~22天) [20]。目前有文献推荐对于不典型川崎病同时又是丙球无反应型,IL-1受体拮抗剂治疗具有良好效果 [21],另外有文献报道先后两次IVIG及激素治疗后川崎病仍合并HLH患儿,英夫利昔单抗具有满意疗效 [22]。这考虑与阻断其病理生理基础有关。在笔者报道的本次病例中,患儿两次丙球冲击治疗无效,在发热病程的第23天发生HLH,比上述文献报道时间要延迟,考虑可能与早期丙球治疗有关,其次积极抗感染治疗也为预防发生HLH争取了时间。但实际上由于家属不配合检查及治疗,未能规范有效激素治疗以早期抑制全身严重炎症反应,可能是导致该病例发展为HLH的至关原因,因此对于丙球无反应川崎病早期激素及免疫抑制剂治疗具有重要作用。
总之,不完全性川崎病如出现肝脾增大、持续高水平血清铁蛋白或持续反复高热等,不但提示其严重全身炎症反应,更可能提示其将发生HLH的倾向。对于该类患儿早期诊断和及时的初步治疗都是预防HLH发生的关键因素。此外,医患之间应加强沟通,互相信任,提高对疾病的认识,才能在疾病诊治过程为挽救生命发挥重要作用。
特别说明:该病例报道已获得病人的知情同意。
参考文献
NOTES
*通讯作者。