1. 引言
在[2,3]重排反应中,根据重排前体的差异可分为两种类型,分别是叶利德型[2,3]重排和阴离子型[2,3]重排(见图1) [1]。作为一类具有代表性的叶利德型[2,3]重排反应,[2,3]-Stevens重排,以烯丙基胺的季铵盐为底物,经碱拔质子后重排得到胺基α位烯丙基化的产物(见图2) [2]。而以苄基胺的季铵盐为底物时,也能实现类似于[1,2]-Stevens重排的反应过程,我们把这类苄胺的重排反应叫做Sommelet-Hauser重排。该反应最初是由Sommelet在1937年发现的 [3],随后,Hauser等对该类反应进行了进一步研究(见图3) [4] [5]。与[1,2]-Stevens重排反应不同的是,S-H重排底物中含有芳环,反应过程中会产生独特的去芳香化中间体,进而恢复芳香性得到胺基邻位芳基化的产物。在S-H重排反应中存在着与之竞争的副反应通道即[1,2]-Stevens重排。造成上述副反应的主要原因是硫鎓盐中间体存在多个脱质子位点,如图所示,反应过程中,碱既可以脱除1号位的质子形成[1,2]-Stevens重排前体,也可以拔除2号位质子得到S-H重排前体(见图4) [1]。因此,S-H重排存在严重的选择性问题,在该反应发现后的几十年时间内都没有太多的后续研究,关于其应用的报告更是少之又少。近些年来,随着新反应策略的出现以及新催化体系的发展,一些新型的S-H重排被相继开发出来,使得该反应又逐渐引起了人们的关注。本文对现有的S-H重排反应的发展进行讨论总结,对重排中断键处的原子种类的差异进行分类,将反应类型分为C-N型、C-S型、以及其他类型(包括N-S型和O-S型)进行讨论。

Figure 1. Nallylic [2,3]-Stevens rearrangement and anionic [2,3]-rearrangement [1]
图1. Nallylic叶利德盐[2,3]-Stevens重排和Anionic阴离子[2,3]重排经典反应
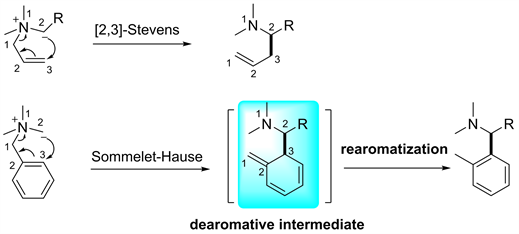
Figure 2. [2,3]-Sommelet-Hauser rearrangement and [2,3]-Stevens rearrangement [2]
图2. [2,3]-S-H重排与[2,3]-Stevens

Figure 4. [1,2]-Stevens and [2,3] S-H rearrangement [1]
图4. [1,2] -Stevens与[2,3] S-H重排
2. C-N型[2,3] S-H重排
2.1. 脱硅C-N键[2,3] S-H重排
由上所述,[2,3] S-H重排从被发现以来,关于它的研究与应用较少,因为这类反应副反应很多,产物选择性差。为了克服这个弊端,1985年Sato和Shirai [6] 小组预合成了一类邻位烷基硅取代的苄基胺季铵盐底物F,利用氟负离子能高选择性的切断碳硅键这一独特性质,使之与氟化铯反应,从而得到相应的重排前体,然后再经由S-H重排得到目标产物。与传统的脱质子策略相比,该方法不需要加碱就可以形成重排前体,因此有效的规避了选择性脱质子的问题,使得[1,2]-Stevens重排得以抑制(见图5)。在该反应中,底物的官能团兼容性好,能够兼容卤素、氰基、酰氧基等有较大衍生价值的官能团(见图6)。
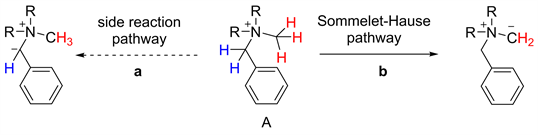
Figure 5. Side reaction of [2,3] S-H rearrangement
图5. [2,3]-S-H重排的副反应

Figure 6. Desilicon C-N type [2,3] S-H rearrangement [6]
图6. 脱硅C-N键[2,3] S-H重排
随后Y. Sato课题组进一步将该脱硅重排策略应用到了脱芳构化反应(图7式1) [7],以及杂芳烃的[2,3] S-H重排中(图7式2) [8]。与此同时,该课题组还开发了脱锡C5-N型S-H重排(图7式3) [9],通过在胺α位引入锡原子,使之与正丁基锂反应,也能以不错的收率得到相应的重排产物。
2.2. N-苄基氨基酸衍生物[2,3] S-H重排
K. Sienkiewicz [10] 在研究N-苄基α-氨基酸或α-氨基酮衍生物的[1,2]-Stevens重排时发现,在某些条件下能够选择性的发生S-H重排,得到了胺基α-苄基化产物。因此,从这一背景出发,Tayama等人猜想能否通过条件的优化(包括不同的碱及碱的形态,反应温度)以及底物的改造,抑制[1,2]-Steven重排,实现高选择性的S-H重排。首先,Tayama等对反应所用的碱的形态及不同的碱进行了大量探索,发现在叔丁醇钾预先溶于THF中,再加入到反应体系中时,能够较好的抑制[1,2]-Stevens重排(见图8) [11]。同时,作者还发现,底物的电子效应和空间效应同样对反应的选择性有较大的影响。吸电子基团在一定程度上促进S-H重排的发生。底物N上取代基较大时,反应更趋向于发生[1,2]-Steven重排。降低温度有利于S-H重排的发生(见图9) [12]。

Figure 7. Other C-N type [2,3] S-H rearrangement [7] [8] [9]
图7. 其它一系列脱硅C-N键[2,3] S-H重排

Figure 8. [1,2]-Stevensor [2,3] S-H rearrangement in presence of different bases [11]
图8. 不同的碱及碱的形态诱导[1,2]-Stevens或者[2,3] S-H重排

Figure 9. [2,3] S-H rearrangement of α-amino acid derivatives [12]
图9. 氨基酸衍生物的[1,2]-Stevens或者[2,3] S-H重排
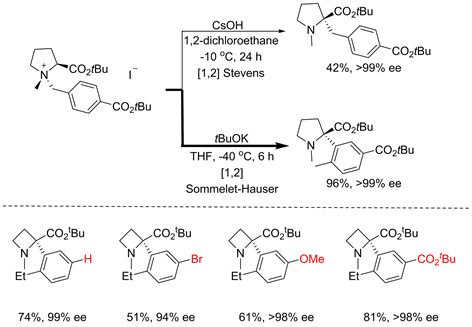
Figure 10. Influence of chiral auxiliary [12]
图10. 手性辅基的对[2,3] S-H重排的影响
受到T. Nakai课题组关于不对称[1,2]-Stevens重排工作的启发 [13],Tayama猜测手性辅基的引入同样也能够诱导不对称S-H重排 [12]。果不其然,简单的改变Nakai反应中的温度以及碱的种类,就能够以优异的产率、高对映选择性的得到S-H重排产物。尽管该反应展现出了一定的官能团兼容性,但是底物骨架仅限于含脯氨酸的季铵盐,单一的结构在某种程度上限制了产物的进一步衍生以及反应的后续应用(见图10)。
为了改变上述困局,Tayama对该反应的手性辅基进行了深入的筛选。作者发现直接在羰基上引入简单的大位阻手性辅基L-薄荷醇即可以较好的产率、优异的立体选择性得到S-H重排产物 [14]。接着作者对该反应的官能团兼容性进行了探索(见图11),发现芳基上连有吸电子基团时能够促进重排反应的进行,一系列吸电子官能团如酯基、氰基、羰基、三氟甲基等都能够很好的兼容于反应体系中,为产物的进一步官能团化提供了一定的空间。令人意外的是,供电子基团的引入使反应无法进行,作者猜测造成这种巨大差异的原因可能是连有供电子基的硫鎓盐中间体不稳定导致的。
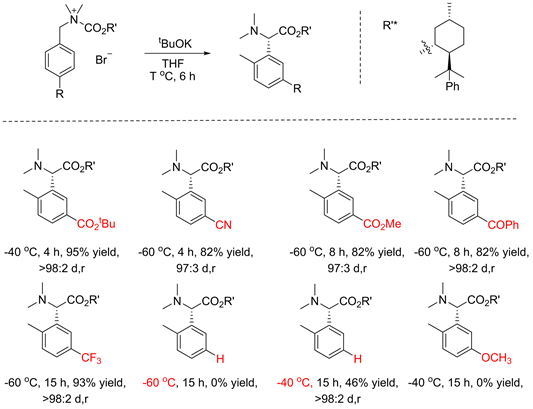
Figure 11. Asymmetric [2,3] S-H rearrangement promoted by chiral menthol [14]
图11. 手性薄荷醇诱导的不对称[2,3] S-H重排
2.3. 苯炔参与的[2,3] S-H重排
2019年,Biju课题组报道了苯炔参与的S-H重排反应 [15]。该反应以叔胺为底物,通过与苯炔反应得到如下图中的A,有趣的是,反应产生的苯负离子能够会拔去季胺邻位碳上的氢,得到[2,3] S-H重排所需的叶利德盐中间体,再经重排得到产物。该反应与传统的[2,3] S-H重排相比不需要加碱拔质子,而是通过分子内质子转移去形成重排中间体,避免了而外添加的碱对反应体系的影响,再在EWG基团的影响下,促进反应朝[2,3] S-H重排方向进行。并且在反应的底物拓展中发现苯炔上连有供电子基团时,能够获得较高的产率,而连有吸电子基团时的产率较低。并且苄基上的苯环在间位上连有小位阻吸电子基团时产率较高,但吸电子基团位阻较大时则仅能得到很低的产率。且在苯环变为富电子芳烃时也仅有较低产率,而且扩展到在杂原子芳烃吡啶环中也有不错的收率(见图12)。
2.4. 氮卡宾金属催化[2,3] S-H重排脱芳构化
2018年顾振华老师课题组报道了通过氮卡宾金属催化[2,3] S-H重排脱芳构化 [16]。反应通过铜催化剂与氮卡宾反应得到金属卡宾中间体,再与叔胺结合得到季胺[2,3] S-H重排中间体,然后经由重排得到去芳香化产物。在反应底物范围上,与氮相邻较近的苯环上连有供电子基团时产率较低,且连有的基团在α位上时,反应不能得到产物,且该反应在羰基上引入手性基团时,产物的dr值为1:1 (见图13)。
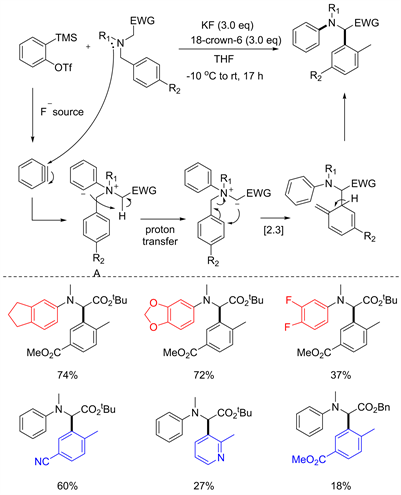
Figure 12. [2,3] S-H rearrangement of benzyne [15]
图12. 苯炔参与的[2,3] S-H重排

Figure 13. Cu-catalyzed [2,3] S-H rearrangement and dearomatization reactions [16]
图13. 铜催化[2,3] S-H重排脱芳构化
3. C-S型[2,3] S-H重排
3.1. 经典C-S型[2,3] S-H重排
经典C-S型[2,3] S-H重排反应类似于传统的C-N型重排,存在着反应步骤较多,以及反应选择性较差的问题。例如1991年Masazumi报道的首例C-S型[2,3] S-H重排(见图14) [17],首先通过付克烷基化反应在芳环上引入硫醚官能团,接着在AgBF4作用下与碘甲烷反应构建相应的硫鎓盐中间体,最后经由碱脱质子重排得到目标产物。该反应一共经历4步,且反应过程中同样需要碱的加入,因此也存在[1,2]-Stevens重排等竞争反应,反应选择性较差。
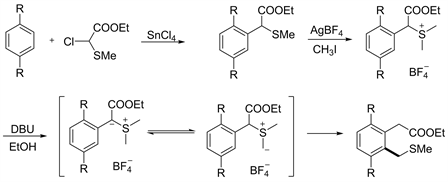
Figure 14. Classical C-S type [2,3] S-H rearrangement [17]
图14. 经典C-S型[2,3] S-H重排
3.2. 杂环芳烃C-S型[2,3] S-H重排
在为数不多的C-S型[2,3] S-H重排中,不得不提到的是1989年Yamada报道的杂环芳烃的重排反应 [18],该反应相较于Masazumi报道的[2,3] S-H重排,步骤相对简当,只需将二甲硫醚气体通入溴代底物中,然后加碱使之重排即可。除了4-酮吡喃类底物之外,该反应还进一步拓展到了呋喃、噻吩、苯并噻吩等杂芳烃化合物中,展现出了一定的普适性(见图15)。
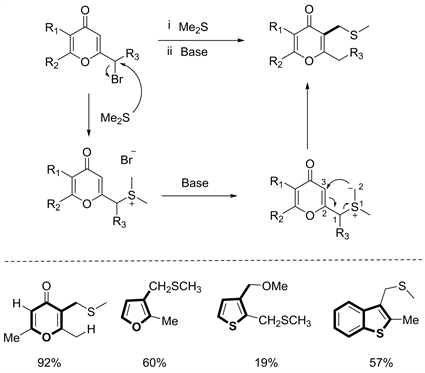
Figure 15. [2,3] S-H rearrangement of heteroaryls [18]
图15. 杂环芳烃[2,3] S-H重排
3.3. 金属催化氮卡宾[2,3] S-H重排
早期的C-S型[2,3] S-H重排需要预先合成硫鎓盐,存在着反应步骤较多,反应通道多,产物选择性较差的问题,所以依旧面临着跟C-N型[2,3] S-H重排一样的困境。有意思的是,2000年Aggarwal [19] 在研究缺电子烯烃的金属催化环丙烷化反应中,存在有[2,3] S-H重排副产物,但没有做深入的研究。于是2008年王建波老师受这个启发,发展了首例金属卡宾与硫醚组装的新反应模式,开启了金属催化S-H重排的新篇章 [20]。该反应首先用氮卡宾与金属催化剂结合形成金属卡宾,然后再与硫醚组装构建重排前体硫鎓盐,然后经过[2,3] S-H重排得到去芳香化的产物,再回复芳香性得到最终产物。该反应一步构建重排前体,使得反应更加的简便,产物的收率在38%~97%之间。再之后的2019年,Koenigs [21] 把杂环芳烃引入到氮卡宾金属催化[2,3] S-H重排中。通过改造硫醚的一端带有杂环芳烃,使得这一反应的适应范围进一步扩大(见图16)。
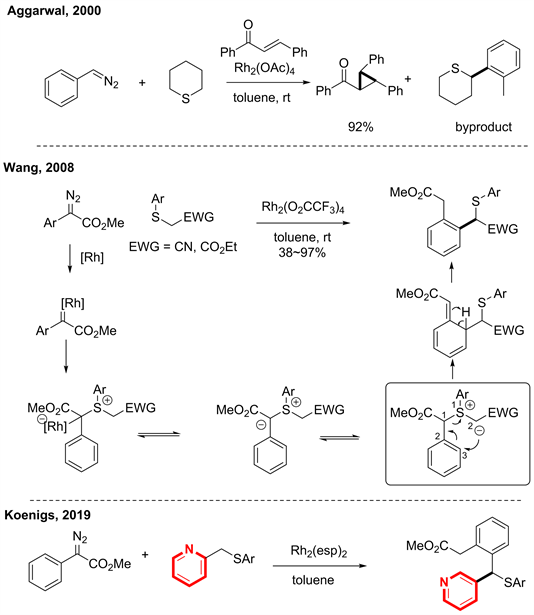
Figure 16. Metal-catalyzed C-S type [2,3] S-H rearrangement reactions [19] [20] [21]
图16. 金属催化的C-S型[2,3] S-H重排
不得不提的是,2019年Koenigs [22] 又进一步提出了氮卡宾金属催化C-Se型[2,3] S-H重排。与上述相同,该反应将S替换为Se,再与金属卡宾(由氮卡宾与金属催化剂反应)组装构建重排前体,然后经[2,3] S-H重排得到去芳香化的产物,再回复芳香性得到最终产物。该反应适用性广,苄基苯环上连有吸电子基团或供电子基团时均能有较高的收率,且反应可以通过控制与Se的α位碳上连接的官能团电性,来控制反应朝着[2,3] S-H重排或者[1.2]-Stevens重排进行(见图17)。

Figure 17. Metal-catalyzed C-Se type [2,3] S-H rearrangement reactions [22]
图17. 金属催化的C-Se型[2,3] S-H重排
3.4. 光激发C-S型[2,3] S-H重排
光激发的反应是一种绿色的引发模式,具有反应条件温和,控制简便等优点,近些年来引起了广泛的关注 [23] [24] [25]。Koenigs在2019年报道了一种新的反应方式,只需通过光激发就能发生[2,3] S-H重排 [26]。反应通过470 nm的蓝光激发氮卡宾,再和硫醚组装形成硫叶立德盐,然后再重排得到产物。并且可以通过控制硫上连接的基团结构来选择性进行[2,3] S-H重排或者[1,2]-Stevens重排。当硫上连有带有吸电子基团的二级碳时,进行[2,3] S-H重排,反应底物范围广,产率也较高。而当和硫连接的基团为二甲酰亚胺时,则进行[1,.2]-Stevens重排(见图18)。
3.5. 不对称C-S型S-H重排
在之前的C-S型[2,3] S-H重排反应中,反应产物不存在手性选择性,导致这一类反应的应用存在局限性。2019年,冯晓明老师课题组基于前期在金属不对称催化反应工作中,开发了重氮吡唑酰胺这一具有广泛底物适用性的手性选择性金属催化剂 [27],该催化剂具有N,N'-二氧化物手性亲核口袋结构,可以以立体选择性与底物结合,使得亲核进攻指向Ni(II)-卡宾中间体,生成S-立体异构中心。将该催化剂引入到 C-S型[2,3] S-H重排反应中,将底物改造为带有吡唑环,使得氮杂环导向基能够与手性路易斯酸键合得到叶利德,从而控制了1,3-质子移位和[2,3] S-H重排,得到所需的立体选择性产物。并对反应的适用性做了深入的研究,在苯环各种官能化化合物和各种大位阻硫醚都具有很高的产率及优异的对映选择性(见图19)。
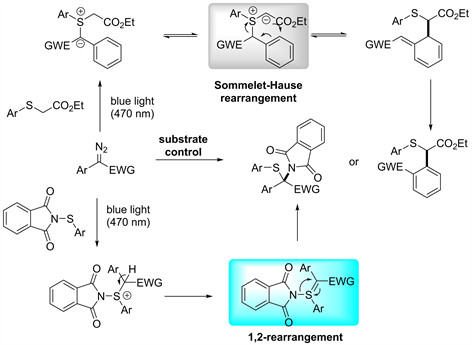
Figure 18. Photo-promoted C-S type [2,3] S-H rearrangement reactions [26]
图18. 光激发C-S型[2,3] S-H重排
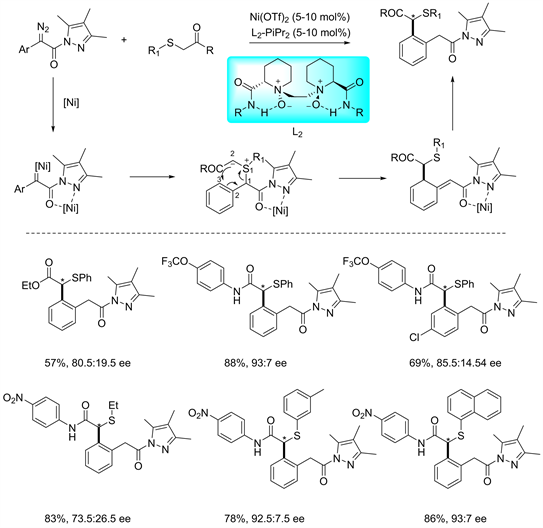
Figure 19. Asymmetric C-S type [2,3] S-H rearrangement reactions [27]
图19. 不对称C-S型S-H重排
尽管反应有较好的产率及手性选择性,但反应的底物受限于吡唑类化合物。这使得反应的应用受到了很大的限制。王剑波老师于2020年报道了唑啉类手性催化剂(如上图L*) [28],该催化剂不需要氮杂环导引即可选择性得到所需立体结构产物。该催化剂可在与铜催化剂共同作用下,以较好的产率及手性选择性得到[2,3] S-H重排产物。反应在苯环上带有供电子及吸电子基团时均能较好的适用(见图20)。
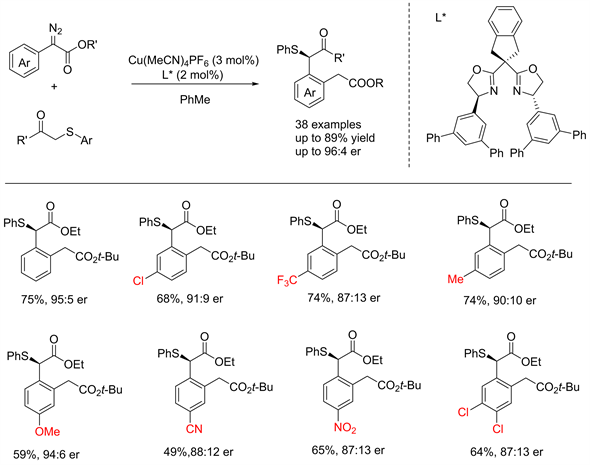
Figure 20. Asymmetric C-S type [2,3] S-H rearrangement reactions without using directing group [28]
图20. 无导向基的不对称C-S型S-H重排
4. 其它[2,3] S-H重排
除了C-S型[2,3] S-H重排和[2,3] C-S型[2,3] S-H重排外,还报道了一些发展较少的X-S型[2,3] S-H重排,其中X杂原子可以是N或者O。
例如Gassman在1973年报道了首例N-S型[2,3] S-H重排,由叔丁氧氯与苯胺反应得到亲电性的氯代苯胺,然后再和硫α位碳上连有吸电子基团的硫醚组装形成叶利德盐,再经过重排得到去芳香化产物,之后再回复芳香性得到具有生物活性骨架的2-吲哚酮类产物 [29]。还有王剑波老师课题组基于2008年对氮卡宾金属催化C-S型[2,3] S-H重排的研究 [30],于2011年报道了首例氮卡宾金属催化N-S型[2,3] S-H重排。通过氮卡宾于金属催化剂反应后再与硫烷基保护的苯胺组装构建C-S型[2,3] S-H重排前体,然后经过重排得到去芳香化产物,再回复芳香性(见图21) [31]。
再有,1965年Kondo报道了首例的O-S型[2,3] S-H重排 [32]。该反应的底物主要是苯酚类化合物。与上述不同的是,二甲基亚砜需要经由DCC活化得到的高亲电性的四价硫后,再与苯酚组装得到O-S型[2,3] S-H重排前体,然后经由重排得到收率为40%~88%的苯酚邻位甲硫基甲基化产物。值得注意的是,1967年Burden报道了O-S型去芳香化[2,3] S-H重排,通过在苯酚的邻位引入两个甲基,以2,6-二甲基苯酚为底物与二甲基亚砜经DCC活化后的产物组装形成O-S型[2,3] S-H重排前体,然后由重排得到甲硫基甲基化去芳香化产物,产物的收率为66% [33]。但由于该反应有数量众多的副产物以及反应时间过度会影响产率及副产物增多的问题,所以这一类反应的应用受到了极大的限制,更待进一步的发展(见图22)。
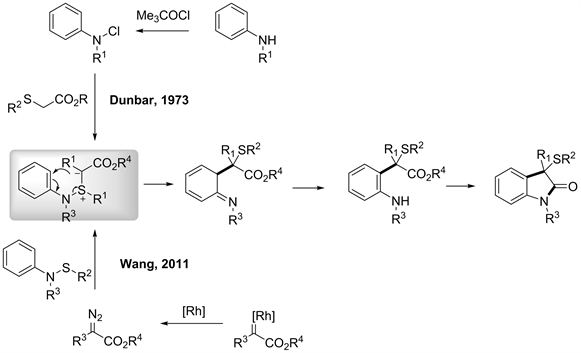
Figure 21. N-S type [2,3] S-H rearrangement reactions [31]
图21. N-S型S-H重排

Figure 22. O-S type [2,3] S-H rearrangement reactions [33]
图22. O-S型S-H重排
5. 结论
经过几十年的发展,这个重排经由[1,2]位的第一选择性发展到了通过反应条件、底物结构以及催化剂来控制选择性地进行[2,3] S-H重排。并且发展出了多样化的组装方式,优化了反应的立体选择性,优化反应步骤的简化,使用更加廉价有效的催化剂等,使得[2,3] S-H重排在底物适应性、反应收率及非对称选择性上都有了不错的进步。但由于还与[1,2]-Stevens重排存在不同点位拔质子导致走向不同的反应通道,以及其它竞争的副反应通道,使得反应的发展应用仍受到副反应较多、产率较低以及受产物空间结构影响情况复杂等限制,期待后来者进一步解决反应过程中存在的问题,让[2,3] S-H重排扩展到更广阔的应用中去。
基金项目
金华市科技计划项目(2018-04-045)。
NOTES
*通讯作者。