1. 引言
1.1. 研究背景
随着数据的爆发性增长以及计算能力的不断提高,人们认识事物的思维方式和研究方法已经逐步从实验观测、理论推演、计算仿真发展为第四种研究新模式——“数据探索” [1]。新模式以数据驱动为主导,建立了数据之间的关系,带来了颠覆性的效果,也是引发社会变革的根本性原因之一。海量数据的到来带来了机遇的同时也在考验着现如今对大数据收集、分析、管理和访问等的处理能力。材料基因工程的提出和发展,促进了多学科交叉领域的发展,也强化了计算机应用范围,形成了材料领域学科的重要发展方向之一。而材料数据作为材料基因工程的三大支撑工具之一 [2],将计算机技术应用于材料领域,针对材料数据的分析和处理过程进行优化,将有利于推进材料领域的科研发展。然而由于实验仪器设备、实验操作人员的习惯以及实验观测环境等因素,针对同一材料会有不同的记录结果,从而导致大量冗余材料数据,因此有必要对冗余数据进行去重,减少其空间占用和计算的成本。
1.2. 研究现状
冗余数据的分析和处理一直都是一个重要的研究课题。一方面,通过冗余数据的分析和处理技术,一定程度上可以提高空间的利用率,另一方面,还可以降低传输及计算的成本。目前,计算机领域上冗余数据的缩减主要通过数据压缩、差分编码以及重复数据删除三种典型的技术 [3]。三种冗余数据的缩减技术本质上都是通过检测冗余数据的方式并采取更短的指针实现数据的缩减。冗余数据的分析处理技术则可以分为两类:一是相同数据的检测技术,包括了完全文件检测技术、FSP技术、CDC技术以及Slidingblock技术等等 [4]。第二类是相似数据的检测和编码技术,可以通过shingle技术、bloomfilter技术或者模式匹配等技术计算指纹相似性 [5],再使用delta编码技术进行数据压缩或直接删除。然而计算机领域中的数据去重更注重于数据物理层面上的相同或相似,而材料科学领域的去重则更注重于数据逻辑层面上的相同或相似。因此,传统领域上的数据去重技术不完全匹配于结构数据的去重,在对结构材料数据进行去重时应当结合材料结构数据特点,对传统数据去重技术进行调整和完善。
冗余材料数据的去除通常包括以下步骤:数据特征提取、指纹计算、数据删除。在材料科学领域的应用上已有多种识别结构相似性的算法,主要从以下几个方面对其指纹进行计算:1) 原子基本信息的描述。包括径向分布信息 [6]、光谱衍射信息 [7]、键合信息 [8] 和力场及键合模式 [9] 等。2) 结构拓扑信息的描述 [10]。3) 结构间点模式映射 [11]。这些指纹计算方式在一定程度上都能表征相应晶体结构并计算出结构间的相似性。但这些算法依旧存在着准确率不高、鲁棒性低、计算效率低下等问题。
2. 基于原子间作用势的晶体结构去重算法设计
2.1. 相关知识
本文中所研究的材料对象为晶体结构,为更好地了解晶体结构的描述方式以及去重算法的原理,首先对涉及的相关知识做简单的介绍:
由于晶体结构具有周期性,一个晶体结构通常使用晶格及晶格内原子的位置进行描述。对晶格的描述通常由三个晶格矢量A、B、C以及中间角度α、β、γ给出,晶格内原子位置则使用笛卡尔坐标或者分数坐标描述。
晶格:将具有一定的原子群使用假想的线连接起来,构成的一个平行六面体的框架,即为晶格。如图1(a)所示。
素晶胞:即基元,是晶体微观空间中最小重复单元。如图1(b)所示。
空间群:空间群指的是晶体内部中全部对称要素的集合。
晶胞:是构成晶体的最小重复单元,包括了晶格、晶格内原子的位置以及空间群三部分信息,用于描述晶体内部的原子和粒子的分布情况,如图1(c)所示。
超晶胞:对晶胞进行扩胞处理,即形成超胞,可认为是对晶胞的扩展。
范德华力:分子间作用力,存在于中性分子或原子间的弱碱性的电性吸引力。可以分为诱导力、色散力以及取向力。
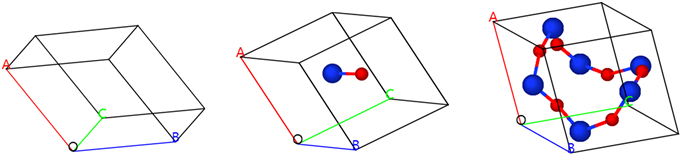 (a) 晶格 (b) 素晶胞 (c) 晶胞
(a) 晶格 (b) 素晶胞 (c) 晶胞
Figure 1. Schematicdiagram of lattice, primitive cell, unit cell
图1. 晶格、素晶胞、晶胞示意图
2.2. 数据特征提取及指纹描述
数据压缩、差分编码以及重复数据删除这三种典型的缩减技术,除了数据压缩技术是以信息论为基础,差分编码和重复数据删除均以数据块为单位分析数据的匹配情况,实现数据的缩减。材料结构数据的缩减与计算机领域的冗余数据缩减技术具有异曲同工之处。首先,它们都需要对数据进行划分,然后对数据块进行指纹计算,通过指纹判断数据的匹配程度,实现数据的缩减。然而,针对材料结构数据,由于实验仪器设备、实验操作人员的习惯以及实验观测环境等因素,会导致针对同一材料会有不同的记录结果,从而导致冗余数据。这一冗余数据在字符串层面是不同的,因此传统的方法失效,需要从数据的物理层面鉴定不同结构数据的相同以及相似程度。
本文提出了一种基于原子间范德华作用势(van der Waals, VDW)的晶体结构的去重算法,旨在提高晶体结构去重算法的适用性,降低计算成本,提高计算效率。算法通过对晶体结构信息进行提取和降维处理,基于原子间相互作用信息,构建晶体指纹,再利用加权余弦距离计算晶体结构间的相似性,最后采用聚类的方法对晶体结构进行去重。图2显示了算法的主要步骤:
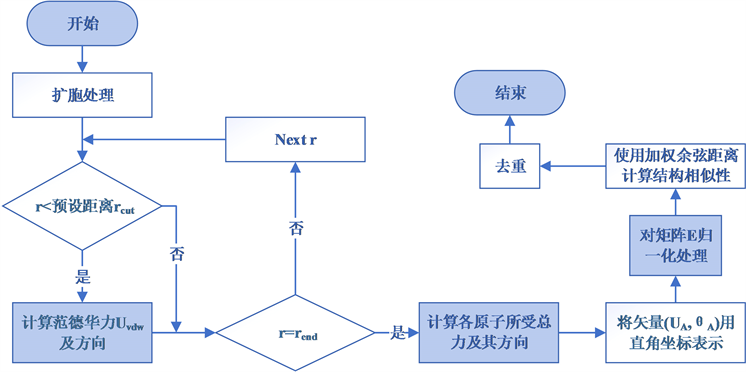
Figure 2. Flow chart of deduplication algorithm based on interatomic potential
图2. 原子间作用势去重算法流程图
步骤一:对晶胞进行扩胞处理,形成一个3 × 3 × 3的超晶胞。
步骤二:对距离小于
的原子对计算原子所受的范德华势
。
步骤三:计算各原子受到力的总和,并用
表示,其中
表示原子A所受原子间作用势的大小信息,
存储原子A所受原子间相互作用的方向信息。
步骤四:将矢量
转换成直角坐标
的表示方式。
步骤五:使用三维矩阵存储各原子的受力信息
,并进行归一化处理。
步骤六:利用加权余弦距离计算两个晶体结构的相似性。结果越接近0,说明结构越相似;结果越接近1,说明结构越不相似。
步骤七:通过聚类的方式对相似晶体去重,其中相似度大于0.75的结果记为两个晶体相似。
详细说明见下文。
晶体结构的相似性检测与文本、文件数据的相同或相似数据的检测不同。晶体结构使用文件的形式记录并存储,一个文件表示一个结构,简单地对结构文档内的数据进行相同或相似检测无法达到结构去冗余的效果。由于实验中的噪音、误差,相同的结构可能会出现不相同的实验数据,对文档数据去重无法高效地做到相同晶体结构的去重。此外,由于材料数据的维度多、数据间关联性高,使用传统的相似度检测的方式容易将不相似的结构误判为相似,造成检测过度的情况。
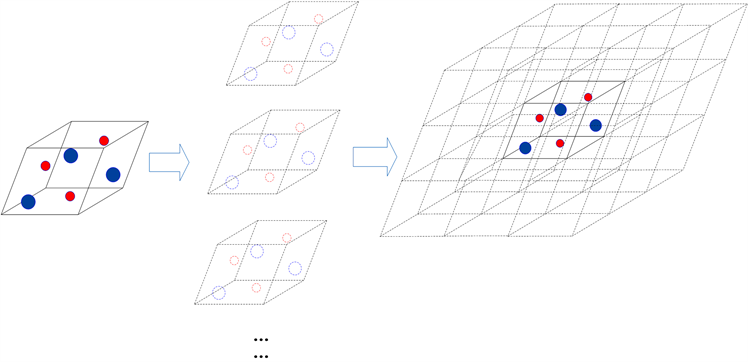
Figure 3. Schematic diagram of cell expansion
图3. 扩胞处理示意图
针对材料结构数据维度多、结构关联性高的特殊性,本文提出的基于范德华势的晶体结构去重算法,对结构数据进行提取、降维,再对数据进行相似度计算、去重。下面对材料结构数据的提取和降维过程进行详细介绍。
该去重算法结合了原子类型、原子间类型、原子能、原子间相互作用力及其方向等因素,旨在提高算法的准确性、适用性和实用性。一个晶体结构由多个相同的晶胞组合而成,而结构文档数据是对晶胞的描述文件。为了范德华势的计算更能准确地表示结构的真实情况,使提取的信息的具备唯一性和真实性。算法首先会根据结构的空间组信息、晶格信息以及原子的相关信息,对晶胞进行扩胞处理,如图3所示,形成一个3 × 3 × 3的超晶胞。
为了降低材料结构的计算成本,该算法还根据材料结构的特性,设置阈值,降低算法时间复杂度,提高计算速度。根据分子作用力与原子间的关系,当原子距离
时,分子作用力几乎为零,其中
处为斥力和吸引力平衡的地方。因此,算法规定仅对
的原子对计算其分子间作用力,如图4所示。
在计算原子间作用力时,使用具较高稳定性的Lennard-Jones 6-12方法进行计算,公式如下:
(1)
计算得到的范德华势的大小和方向,使用矢量
进行描述,其中A为晶胞内的原子,B为扩胞内的原子。
,
,
为原子A的范德华原子能,
为原子A的范德华距离,
为原子A和B间的距离,
,
表示A原子与B原子范德华势的大小,
表示范德华势的方向,A、B原子则为两个不同的原子。
随后通过计算各原子的势能之和,记录晶胞内各原子受到的范德华势信息。根据矢量求和的方式,计算各个原子范德华势的和,如图5所示,并用
表示。其中
表示原子A所受范德华势之和的大小,
表示原子A所受范德华势之和的方向,使用向量
表示。
为统一格式并便于后续计算,将
的表达方式使用直角坐标描述,表示为
,其中:
(2)
(3)
然后将每个原子的信息
整合,并用三维矩阵
进行描述。
到这里,已经将晶体结构的信息进行降维、提取。将描述晶体结构的文档信息提取至一个三维矩阵中,所得到的矩阵
即可唯一代表一个晶体结构信息。算法解决了结构数据维度多、结构关联系高、无法直接通过对文本信息检测相同或相似数据的问题。成功将文档数据的检测对比转换为数学问题,算法仅仅需要分析矩阵信息,便可判断两个结构是否相似。
但是由于对结构的表示不统一,标准不一的问题,各个结构间无法直接进行比较或者计算他们的相似性,因此,还需要对矩阵
进行“归一化”处理,得到矩阵E,矩阵E即用于表征该晶体结构。
2.3. 指纹相似度计算及删除
对晶体结构信息进行提取、降维以后,需对晶体结构进行相似度计算。由于材料数据的影响因素比较复杂,各个因素的影响程度不一,同时考虑到各原子受到的范德华势的角度的影响,该算法使用加权余弦距离计算结构间的相似度。两个结构对应的原子A、
两个原子的相似度计算公式为:
(4)
(5)
类似,则两个结构的相似性计算公式为:
(6)
(7)
距离D即为用于描述两个结构的相似程度的标准,当
的值越接近0,则表示两个晶体结构的差异程度越高;当
的值越接近1,则表示两个晶体结构越不相似。
最后,根据实验经验,本文对于相似度大于0.75的结果记为两个晶体相似,通过聚类的方式对相似晶体去重,保留其中一个最新发表的晶体结构。
3. 性能评估
为了说明基于van der Waals (VDW)势的晶体结构去重算法的性能,本文选取了几个比较经典以及创新性较好的晶体结构去重算法进行复现,与本文提出的算法进行比较实验。所选取的算法如下:
1) 径向分布函数算法(Radial Distribution Function, RDF):算法于1998年提出,是最早提出的也是最为经典晶体结构的去重算法,后来该算法也经过改善优化使得性能大幅度提高,本文复现实验参考MAISE软件 [6] 相似度计算方法,根据粒子在空间的分布机率计算结构建相似性。
2) 键特征矩阵算法(Bond Characterization Matrix, BCM) [8]:算法利用键表矩阵存储键信息,包括键矢量、键角、键类型、键长、键数目信息,用以表征整个晶体结构。该算法对晶体结构的键信息着重描述提取,以此表征整个结构。该算法详细地对某一因素的信息进行提取,具有一定的代表性。
3) 图论算法(Graph Theory, GT):算法使用结构的拓扑信息表征整个晶体结构,每一种晶体结构对应了唯一一个或几个拓扑结构,通过对比其中的拓扑结构,判断两者是否为同一晶体结构。该算法于2019年提出 [10],是具备创新性的方法之一,选取该算法进行复现并对比具有代表性。
表1对各个算法进行了简单描述。为了说明各种算法的优缺点,实验中选取了部分最具代表性的晶体结构进行对比实验。图6给出了部分晶体结构图,结构原子由不同颜色和大小表示。
实验中选取了12个晶体结构数据,8种晶体类型进行对比实验,分别编号为S1、S2、P1、P2、E1、K1、G1、O1。由于GT算法描述结构中原子间的连接情况,通过直接对比拓扑信息图判断结构是否相同,其相似度使用0和1表示,0表示不相同,1表示相同。而RDF算法、BCM算法以及VDW算法给出0到1范围内连续的打分值,其结果越接近1表示该结构越相似。
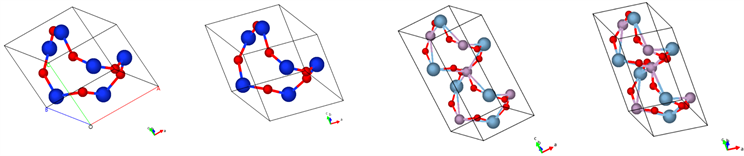 (a) S1(b) S2 (c) P1 (d) P2
(a) S1(b) S2 (c) P1 (d) P2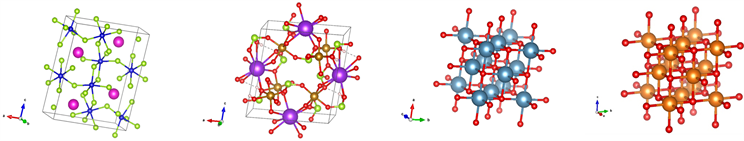 (e) E1 (f) K1 (g) G1(h) O1
(e) E1 (f) K1 (g) G1(h) O1
Figure 6. Part of crystal structure
图6. 部分晶体结构图
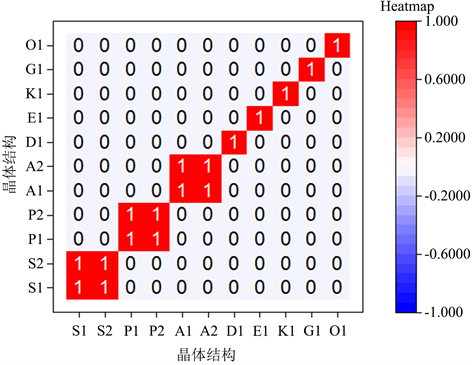
Figure 9. The result of the VDW algorithm
图9. VDW算法的计算结果
3.1. 准确性评估
准确性是评价一个算法性能的核心的指标。准确性通常用于描述实验中的某一实验指标或数据与真值的接近程度。本文选择了12个典型结构对算法的准确性进行评估,用于判断算法计算结果是否正确,能否应用于实际应用中。根据相似度的计算结果,VDW算法将相似度大于0.75的结构对判定为结构相同。然后使用聚类的方式,可以对晶体结构进行去重,得出结果如图7所示。
分别使用GT算法、BCM算法、RDF算法以及VDW算法对数据进行相似性计算,并判断结构是否相似。经统计,可得各算法的去重准确率如图8。其中VDW算法的计算结果可见图9,“1”表示两个结果相同,“0”为不同。
分析统计结果,BCM算法、RDF算法以及VDW算法均能有效的识别结构是否相同,能有效的对冗余结构进行去重。而GT算法由于仅根据各原子见的连接情况进行判断,导致无法正确判断两个原子类型不同而原子连接情况相同的原子是否相同,例如编号为G1和O1的晶体结构。
3.2. 鲁棒性评估
鲁棒性通常用于描述一个算法能否在系统存在一定的不确定的扰动下,维持其性能不变的特性。一个算法是否具有鲁棒性,是算法能否应用于实际情况的关键,是算法的一个最重要的设计指标。为了探讨各算法的鲁棒性,本文首先从算法在不同结构上的计算结构进行了相似度计算,研究算法的通用性。图10~13分别给出了算法GT、RDF、VDW以及BCM的结构相似度结果热力图(具体数据可见附录)。热力图的横纵坐标表示结构编号,表格中
的数据表示第i个结构与第j个结构的相似度,颜色越深表示结果约接近1,两个结构越相似。同理,颜色越浅表示结果越接近0,两个结构越不相似。
从图中可以看出,GT算法仅有0和1这两个计算结果,它仅能用于判断任意两个结构的相同与不相同,而无法计算结构间的相似度。RDF算法、BCM算法以及VDW算法计算结果的域值为[0, 1],能计算结构间的相似度。但是由于BCM算法的特殊性,该算法不能对不同原子类型和数量的结构进行相似度计算。因此,相比之下,RDF算法以及VDW算法可以对任意两个晶体结构的相似度进行计算,从通用性的角度展现了算法的优势,具有更强的鲁棒性。

Table 2. The indicator of algorithm stability
表2. 算法稳定性指标
进一步,本文从稳定性的角度研究算法的鲁棒性。使用如下计算公式(8)对RDF算法与VDW算法稳定性对比。其中,
为算法的稳定性数值,
表示
结构与
结构的相似度,
与
为两个几近相同的结构。
(8)
计算结果可见表2,VDW算法稳定性指标达到0.9654,比RDF算法高出了0.0178。实验表明,VDW算法具有更好的稳定性。综合通用性和稳定性的研究结果,VDW算法的鲁棒性性性能更好。
3.3. 计算效率评估
随着数据量的指数增长,人们对计算效率的要求也逐渐提高。为说明VDW算法的计算效率,本论文通过实验统计了各算法平均每个结构的处理时间,结果如图14所示:

Figure 14. Average computation time of each algorithm
图14. 各算法完成计算的平均时间
结果显示,GT算法在计算效率上更具优势,用时更短,这一结果是符合预期的。产生这一现象的主要原因是由于GT算法的时间复杂度更低,为O(n2)。另外,虽然VDW算法、BCM算法以及RDF算法的时间复杂度均为O(n3),但是在计算效率上,VDW算法仍然比其他两种算法快,分别快了0.0185s和0.0651 s。这是因为VDW算法在提取特征数据时,根据物理知识,设置了阈值,减少了计算成本。BCM则在计算结构相似度前对结构进行了分类和筛选,减少了不必要的计算。而RDF算法则是基于全局的,对结构内的任意原子进行了计算,无疑会导致计算效率的降低。尽管VDW算法和BCM算法分别通过设置阈值、筛选部分结构提高计算效率,但是VDW在计算效率上仍然占优势,体现了VDW算法具有较高的计算效率。
3.4. 应用效果
为了证明VDW算法的可行性,本文从ICSD、CSD和COD三个数据库分别收集了20.8万、97.3万、41.3万,共159.4万数据进行研究。
根据数据的特点,我们将数据分为有机晶体数据以及无机晶体结构两种晶体结构进行处理。由于数据中存在原子占位数小于1.0的结构情况较复杂,需要进行特殊处理,所以本文暂不考虑该类结构。
1) 有机晶体结构的处理
CSD数据库是有机晶体结构的数据源。从CSD数据库中收集了973,631个晶体结构。在通过数据处理,得到了316,380个数据。处理过程如图15所示。
第一步,去除原子占位数小于1.0的晶体结构。对于可以提取其占位数的结构,删除原子占位数小于1.0的晶体结构数据。对于无法提取其占位数的结构,将其占位数默认为1.0,然后去除含有金属元素的结构。至此,本文去除了532,292个晶体结构数据。
第二步,去除无序结构。若晶体结构中存在两个原子间距离
与原子对应的范德华半径和
之比小于0.5,即
时,该结构即为disorder结构,需要时去除。经过这一步,本文去除了42,153个晶体结构数据。
第三步,对结构数据进行溶剂检测,去除其中的溶剂分子。这里去除了50,695个晶体结构数据。
第四步,调用第三章提出的VDW算法,对相同或者相似的晶体结构计算其相似度,进行去重,并根据数据的更新情况,选择最近发表的晶体保留,去除了32,111个晶体结构数据。
2) 无机晶体结构的处理
无机晶体结构数据主要从ICSD数据库以及COD数据库中获取。本文从ICSD数据库中收集了208,425个晶体结构数据,从COD数据库中收集了413,357个晶体结构。经过数据的处理与合并后,最终获得了92,791个结构数据。具体处理过程如图16所示。
由于ICSD数据库和COD数据库的数据存储结构不同,本文在预处理的过程时,分别对两种数据进行处理。与有机晶体结构的处理过程相同,首先去除原子占位数小于1.0的晶体结构数据,此时ICSD数据库剩余116,335个晶体结构,COD数据库剩余281,400个晶体结构。此外,由于COD数据库中存在部分有机晶体结构,本文将结构中存在C-C键和C-H键的结构认定为有机晶体结构,去除有机晶体,最终获得45,988个晶体结构。
由于无机晶体结构数据的复杂性较高,无机晶体结构数据的去重计算量过大,为减少计算量,本文分别对ICSD数据库以及COD数据库使用VDW算法去重处理后将数据合并,并再次去重,最终获得了92,791个晶体结构。
实验证明,无论是针对有机晶体结构还是无机晶体结构,基于原子间相互作用势能的晶体结构去重算法都能在实际场景中有效应用,该算法具有实际应用效果。
4. 总结及展望
本文以材料数据为研究对象,针对传统材料结构相似数据去冗余算法效率低下、数据质量低等问题,提出了基于原子间作用势的晶体结构去重算法。实验结果表明该算法能有效识别相同以及相似晶体结构并去重。与已有算法比较,该算法在保证算法准确性的同时,其鲁棒性、计算效率均占有优势。通过对材料领域ICSD、COD以及CSD超过159万无机以及有机晶体结构数据进行相似度计算,有效去重了10.16万个结构构建了去冗余的晶体结构数据库,相关数据发布于网站:https://matgen.nscc-gz.cn/。
基金项目
广东省重点领域研发计划(2019B010942001);广东省引进创新创业团队项目(2016ZT06D211)。
附录
Table A1. Results of RDF algorithm
表A1. RDF算法结果
Table A2. Results of BCM algorithm
表A2. BCM算法结果
Table A3. Results of GT algorithm
表A3. GT算法结果
Table A4. Results of VDW algorithm
表A4. GT算法结果
NOTES
*通讯作者。