1. 引言
DNA用于分子生物学 [1] [2] [3] [4] [5]、基因重组 [6] [7] [8]、酶工程等领域 [9] [10] [11],这些领域十分关注DNA纯度、浓度和总量,所以DNA制备方法至关重要。DNA制备方法有苯酚、氯仿和异戊醇(25:24:1, pH 8.0)抽提–乙醇沉淀 [12],十六烷基三甲基溴化铵(CTAB) [13],氯化铯(CsCl)梯度离心等方法 [14] [15] [16]。经典的CsCl离心最早用于制备质粒DNA,但是需要异丙醇抽提去除溴化乙锭(EB)、乙醇沉淀DNA,透析去除CsCl等多步繁琐工作 [17] [18] [19]。目前,DNA制备采用琼脂糖电泳–胶回收试剂盒、PCR产物柱回收试剂盒 [20]。因为快速方便,胶回收、柱回收成为常用DNA制备方法 [5] [14] [21]。但是,胶回收、柱回收只能满足体外重组DNA小批量制备,难以制备大量–高纯–高浓度DNA,对于大分子量的载体DNA更是如此。
然而,DNA重组常需要大量–高纯–高浓度DNA。外切酶VIII截短体tExo作为胞外同源重组酶时活性检测需要10,000 ng的5.0 kb线性化载体pGEX-6P-1 [8]。ET作为胞内同源重组酶时需要转化高达100 ng/μL目的DNA、浓度高达200 ng/μL的2.2 kb线性载体p15A-cm、10,000 ng/μL的8 kb线性载体pBeloBAC11、及10,000 ng/μL的7.5 kb线性载体pBAC2015 [12]。此类线性载体DNA仍需要苯酚、氯仿和异戊醇、异丙醇沉淀等多步繁琐工作。如果探讨基因重组效率与酶量、反应时间、宿主细胞活性、酶促动力学等多种因素的关系,则需要更大量–高浓度DNA。DNA连接反应、酶切反应中间物通过胶回收纯化时,DNA浓度常低于5 ng/μL,其A260/A230纯度指标只有0.45~0.29。但是,DNA测序浓度要求达到10 ng/μL,纯净DNA 260/A230指标要求达到2。柱回收不能去除未完全反应的底物DNA。基因组编辑时,需要转化达到50,000 ng guide-RNA、50,000 ng Cas9表达到质粒(13.2 kb)、50,000 ng供体DNA (5.2 kb) [22],所以,迫切需要大批量–高纯–高浓度DNA。
本研究基于CsCl梯度离心能够大量制备DNA、柱回收能够快速制备DNA的优势,开发了CsCl离心–柱回收方法。以5.9 kb的PCR产物pET28a-xyn (黑曲霉GH11木聚糖酶基因)为材料 [23],先通过CsCl梯度离心、注射器回收目的DNA、柱回收去除EB和CsCl,从而快速制备大量–高纯–高浓度DNA。进而通过非等量引物PCR优化、6个柱回收进一步提高DNA纯度、浓度、回收量。同时与PCR产物经胶回收、柱回收方法进行比较。
2. 材料与方法
2.1. 材料
Q5 DNA聚合酶,dNTPs,限制性内切酶DpnI、Xho I,T4 DNA连接酶(T4lig)均来自NEB公司(北京)。引物JFV2810 (CAGCCATATGATGAGTGCCG)和JRV2812 (CATATGGCTGCCG CGCGGCACC) (下划线为10 bp同源臂)、DNA回收试剂盒来自于上海生物工程有限公司(上海)。质粒pET28a-xyn (Aspergillus niger GH11木聚糖酶基因)由本实验室构建。
2.2. DNA扩增
PCR扩增体系含有12 ng质粒 pET28a-xyn为模板,250 μmol JFV2810,250 μmol JRV2812,200 μM dNTPs,1 U Q5 DNA聚合酶,1 × Q5 DNA聚合酶buffer,以水补足50 μL,扩增2000 μL PCR产物。PCR程序:98℃变性3 min,28个循环:98℃变性30 s,双退火程序(67℃退火15 s,58℃退火15 s) [24],72℃延伸3 min 45 s,72℃延伸10 min。DpnI消解模板反应体系为:17 μL PCR产物,1 × DpnI buffer,1 μL 20 U/μl DpnI,37℃消解模板4 h,80℃灭活DpnI 30 min。
2.3. CsCl密度梯度离心–柱回收方法
CsCl密度梯度离心:2000 μL PCR产物加水补足4 mL,加入终浓度1.55 g/mL CsCl,30℃下溶解,再加入终浓度0.2 mg/mL EB (溴化乙锭),30℃下溶解。用液体石蜡油加满离心管,热熔器封口。样品在离心机NVT100转子中75,000 rpm超速离心6 h (Beckman, USA),与pET28a-xyn质粒DNA平行超离心作为对照,从而确定目的DNA位置(在302 nm紫外光下观察),用1 mL注射器从离心管顶部缓慢吸出目的DNA至1.5 mL干净EP管中。
柱回收DNA:注射器收集目的DNA溶液中含有1.55 g/mL CsCl和0.2 mg/mL EB,需要去除CsCl和EB得到纯化DNA,根据PCR产物柱回收试剂盒指南 [20],向DNA溶液中加入5倍体积B3缓冲液混匀,而后移入回收柱,8000 g离心30 s,加入500 μL 清洗液(wash solution),9000 g离心30 s,倒掉清洗液,重复清洗一次。回收柱在9000 g离心2 min,在烘箱15 min晾至无酒精味。回收柱膜中央加入35 μL elution buffer,60℃水浴2 min,9000 g离心2 min,得到DNA溶液,探讨1~4次洗脱DNA回收量、浓度、纯度。用Nanodrop 1000检测DNA浓度(Thermo scientific, USA),电泳检测DNA条带。
非等量引物PCR优化:因为引物JFV2810和JRV2812具有10 bp同源臂,PCR反应体系中加入降低10倍的JFV2812引物进行非等量引物PCR扩增 [24],其它PCR试剂和PCR程序如上。探讨2000 μL非等量引物PCR产物经CsCl-EB离心、1个柱回收DNA的效率。
6个柱回收DNA:当DNA量大于回收柱最大承载量时,1个柱回收可能丢失部分DNA,为了提高DNA回收率,探讨了3000 μL非等量引物PCR产物经CsCl-EB离心、6个柱回收DNA的效率。
2.4. 胶回收方法比较
为了与CsCl离心–柱回收方法比较,取2000 μL PCR产物经DpnI消解模板,凝胶电泳后,胶回收纯化DNA,每次洗脱体积30 μL,共洗脱三次,将各批次洗脱液混匀。
取7568 ng质粒DNA用于4份XhoI酶切反应,每份酶切体系含有1892 ng pET28a-xyn质粒,2 μL 20 U/μL的Xho I,1 × cut smart 缓冲液,37℃酶切2 h,XhoI酶经70℃灭活20 min,凝胶电泳后,胶回收DNA。
2.5. 柱回收方法比较
为了与CsCl离心–柱回收方法比较,取2000 μL PCR产物经DpnI消解模板,分别上5个柱回收DNA,每次洗脱体积30 μL,共洗脱三次,将各批次洗脱液混匀。
1768 ng DNA用于4份连接反应,每份连接体系含有442 ng线性质粒pET28a-xyn DNA,1 × T4 DNA 连接酶buffer,400 U T4 DNA连接酶,加水补足50 μL。25℃连接3 h,T4 DNA连接酶经80℃灭活2 h。连接产物经柱回收纯化,依次进行二次连接、柱回收纯化、再经三次连接,而后凝胶电泳检测。
3. 结果与分析
3.1. CsCl密度梯度离心–柱回收方法
2000 μL PCR产物经CsCl-EB超离心后,根据质粒DNA对照中线性DNA位置(图1_3),用注射器回收PCR产物样品中目的DNA (图1_4),凝胶电泳检测显示回收产物为5.9 kb目的DNA (图1_5,6)。注射器回收DNA溶液中含有1.55 g/mL CsCl和0.2 mg/mL EB,经过1个柱回收去除CsCl和EB [20],一次洗脱得到10.8 μg DNA (图1),可以满足10,000 ng线性化载体需要 [8]。回收DNA浓度高达145.2 ng/μL,能够满足100 ng/μL浓度目的DNA的需要 [12],A260/A230纯度指标高达2.35 (表1),达到纯净DNA的A260/A230指标2。经过四次洗脱共得到22.1 μg DNA,四次洗脱得到DNA浓度为61.7 ng/μL,远高于DNA测序要求。
DNA洗脱曲线显示2~4次洗脱DNA量依次减少(图1_8),DNA洗脱曲线表明一次洗脱DNA占DNA总量49%,但是一次洗脱DNA浓度为总DNA的2.1倍,四次洗脱得到22.1 μg DNA,接近于洗脱曲线理论值19.7 μg。目前无论胶回收还是柱回收DNA时,常用一次洗脱,i.e.,回收DNA只有总DNA的一半。该结果表明四次洗脱可以提高DNA总量,一、二次洗脱可以提高DNA浓度。DNA洗脱曲线与异丙醇去除EB效率、酶分子热失活曲线相似 [23],均为指数衰减曲线。
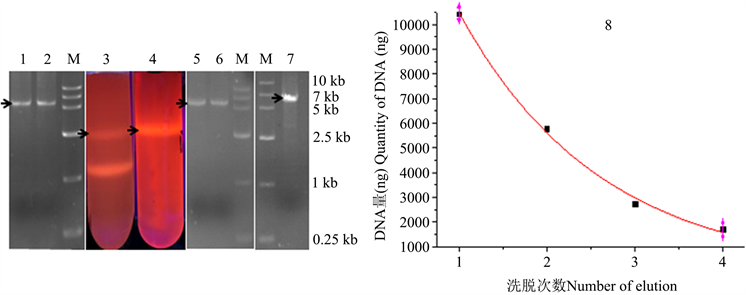
Figure 1. DNA Electrophoresis and CsCl centrifugation, M. DNA marker VI; 1~2. PCR products; 3~4. CsCl centrifugation and ultra-violet assay of plasmid and PCR products; 5~6. Syringe recovery products of CsCl centrifugation; 7. Column recovery DNA; 8. DNA extraction profile
图1. DNA电泳及CsCl离心,M. DNA marker VI;1~2. PCR产物,3~4. CsCl离心和紫外光检测质粒和PCR产物,5~6. CsCl离心后注射器回收产物;7. CsCl离心–柱回收DNA;8. DNA洗脱曲线

Table 1. DNA recovery and quality assay
表1. DNA回收量及质量检测
3.2. 方法优化
非等量引物PCR优化:DNA回收量与PCR扩增效率有关,PCR扩增效率受到同源臂引物影响 [24] [25],所以进行单侧引物量降低10倍的非等量引物PCR扩增。2000 μL非等量引物PCR产物经CsCl-EB离心后、1个柱回收一次洗脱得到17.1 μg DNA,浓度高达189.6 ng/μL,四次洗脱回收DNA高达36.2 μg,是2000 μL常规PCR产物回收量的1.6倍(表1)。
多柱回收:当DNA量大于回收柱最大承载量时,1个柱回收可能损失DNA。取3000 μL非等量引物PCR产物经CsCl-EB离心后、6个回收柱回收,一次洗脱得到28.2 μg DNA、浓度高达391 ng/μL (表1),能够满足对200 ng/μL线性载体DNA浓度的需求 [12],如果要满足10,000 ng/μL线性载体的要求,则需要9000 μL非等量引物PCR产物经6个柱一次洗脱回收DNA。四次洗脱回收DNA量高达61.5 μg,是1个柱回收DNA量的近2倍,说明多个柱回收更利于大量DNA制备。
3.3. 胶回收方法比较
2000 μL PCR产物胶回收得到9.6 μg DNA (只有CsCl离心–柱回收等量PCR产物得到DNA的43%),其A260/A230指标只有0.29 (远低于CsCl离心–柱回收DNA的纯度1.99) (表1)。1.2 μg线性pET28a-xyn DNA经E588酶切产物胶回收浓度只有5.4 ng/μL,达不到DNA测序要求,其A260/A230指标只有0.09,显示DNA中有较多碳水化合物、胍盐污染,因为DNA回收试剂盒使用碳水化合物硅胶,当DNA浓度低时污染较高。
7568 ng 质粒pET28a-xyn经XhoI酶切后产物,胶回收得到1.8 μg DNA,浓度23.3 ng/μL (表1),其A260/A230指标为1.07 (低于CsCl离心–柱回收DNA的纯度1.99),显示有较好的纯度(图2_1,2),但是回收率只有23%。
3.4. 柱回收方法比较
2000 μL PCR产物经柱回收得到66.9 μg DNA,浓度为167.3 ng/μL,其A260/A230指标2.14,可以计算出PCR产物中DNA浓度为0.03 ng/μL。因为柱回收无法去除非特异性DNA条带,凝胶电泳检测柱回收产物显示很多非特异性条带(图2_6,7),与CsCl离心–柱回收DNA纯度有很大差距(图1_7)。
1.8 μg DNA经过第一次连接反应电泳检测有明显产物条带(图2_3),一次连接产物柱回收纯化后经第二次连接反应,电泳检测只有隐约产物条带(图2_4),二次连接产物经柱回收纯化后再经第三次连接反应,电泳检测没有产物条带(图2_5),说明DNA连续柱回收效率很低。凝胶电泳检测的最低限度为10 ng,DNA多步连接反应时,柱回收效率严重影响实验结果,需要高达37.8 μg的DNA用于第一次连接、柱回收,而后用于第二次连接、柱回收,才能经第三次连接后达到电泳检测的限度,所以迫切需要大量–高纯–高浓度DNA。
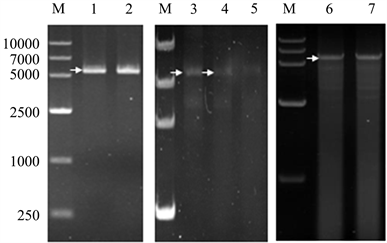
Figure 2. Electrophoresis of column-recovery products, 1~2. Gel-recovery product of XhoI-cut plasmids pET28a-xyn; 3. 1st ligation products; 4. 2nd ligation products; 5. 3rd ligation products; 6~7. Column-recovery of PCR products
图2. 柱回收产物电泳,M. DNA Marker VI;1~2. XhoI酶切质粒pET28a-xyn胶回收产物;3. 一次酶连产物;4. 二次酶连产物;5. 三次酶连产物;6~7. 柱回收PCR产物
4. 讨论
DNA制备大体分为传统方法和试剂盒回收方法 [12] - [18] [26],经典CsCl离心方法最早用来制备质粒DNA [18],但是步骤繁琐,需要异丙醇抽提去除EB、乙醇沉淀DNA [19],透析去除CsCl等操作 [20] [21]。胶回收、柱回收试剂盒方便快速,但是只能制备小量DNA。检测DNA酶学性质往往需要nmol、甚至μmol级别的DNA。本研究基于CsCl离心大量制备和柱回收快速制备DNA的优势,开发了大量–高纯–高浓度DNA的制备方法。3000 μL非等量引物PCR产物经CsCl-EB离心后、6个回收柱回收,一次洗脱得到DNA浓度高达391 ng/μL (表1),能够满足胞内同源重组对浓度高达200 ng/μL线性载体DNA的需求 [12],四次洗脱回收DNA量高达61.5 μg,据笔者所知这是目前达到的最大DNA回收量、纯度、浓度。由表1可知CsCl离心–柱回收DNA A260/A230纯度在1.99~2.35之间,与纯净核酸指标一致,而胶回收DNA A260/A230纯度只有0.29,柱回收DNA则含有较多杂带DNA。CsCl离心–柱回收DNA A260/A280指标与胶回收、柱回收没有太大差别。通过胞外分子内同源重组构建无缝质粒时 [5],12 bp分子内同源臂线性DNA pET21a-XynB4需要360 ng经Exnase II重组得到6个转化子,经外切酶VIII全酶和T蛋白重组得到2个转化子,同样需要大量–高纯–高浓度DNA。
根据CsCl梯度离心平衡时间分离大小不同的DNA。因为大小不同的DNA分子需要不同的离心时间达到平衡,5.9 kb大分子DNA需要6 h离心达到平衡,而185 bp的小分子DNA需要15~25 h离心才能达到平衡,DNA分子量越小所需离心时间越长。这是因为DNA片断越小插入的EB量越少,则其密度越小,所需离心力越高、离心时间越长。DNA需要2 × 105 g的相对离心力,RNA需要4 × 105 g的相对离心力,单链DNA与RNA相似,如果用单侧引物PCR扩增DNA,则PCR产生可能含在单链DNA、非特异双链DNA杂带,通过控制离心力和离心时间去除杂带。
因为需要柱回收去除CsCl离心产物中的CsCl和EB,柱回收试剂盒效率也是影响DNA回收效率的因素。不同公司的试剂盒回收率明显不同 [5] [14],Genemark柱回收率为80~95%,胶回收率为60%~90%,Tiangen回收率为80%。笔者通过胶回收5.9 kb大分子DNA A260/A230指标只有0.29 (表1),回收7600 ng DNA时A260/A230才能达到1.07 (表1),最好使用回收效率好的试剂盒。
5. 结论
综上所述,本研究结合CsCl梯度离心大量制备DNA、柱回收快速制备DNA的优势,开发了大量–高纯–高浓度DNA的制备方法。在此基础上,通过非等量引物PCR扩增、6个回收柱回收进一步提高DNA制备效率,一次洗脱回收DNA浓度高达391 ng/μL,四次洗脱回收DNA量高达61.5 μg,A260/A230纯度指标高达1.99,各项指标均显著高于胶回收、柱回收,从而满足对大量–高纯–高浓度DNA的质量需求。
基金项目
国家自然科学基金面上项目“基于同源/非同源结构置换的木聚糖酶–葡聚糖酶结构元件功能解析”(31771915)。
NOTES
*通讯作者。