1. 介绍
非编码RNA (Non-coding RNA, ncRNA)是指不参与编码产生蛋白质的RNA,这些RNA曾被认为是无用的,长链非编码RNA (Long non-coding RNA, lncRNA)是长度大于200个核苷酸的非编码RNA,越来越多的研究表明尽管不能直接编码为蛋白质,长链非编码RNA仍可以参与一系列生物学过程,例如遗传表现调节、肿瘤生长、免疫反应等 [1]。因此了解并识别潜在的长链非编码RNA-蛋白质关联是很有必要的。虽然现在已有大规模实验方法可以确定两者的关联,但这些实验往往会花费大量的时间和物质成本。如今,研究人员已经将机器学习及深度学习方法用到了预测两者关系上,并取得了优良结果。
近些年来,利用机器学习方法来预测长链非编码RNA与蛋白质相互作用(lncRNA-protein interaction, LPI)已经取得了丰硕的成果,Pan等人 [2] 在2016年提出的一种基于序列信息的利用堆叠式自动编码器来进行预测LPI的模型,称为IPMiner。随后,Xiao等人 [3] 在2017提出了一种整合异构网络使用HeteSim评分来预测LPI的模型,称为PLPIHS。在2018年,Hu等人 [4] 使用线性集成策略集成了支持向量机(Support Vector Machines, SVM)、随机森林(Random Forest, RF)和极端梯度增强(eXtreme Gradient Boosting, XGB)三种模型,并且使用了由三种不同方法提取出来的序列特征,构建了HLPI-Ensemble模型。2019年,Zhan等人 [5] 提出了一个名为BGFE的基于序列的方法来预测非编码RNA与蛋白质相互作用,该模型将堆叠自动编码器网络与随机森林分类器相结合,并采用了奇异值分解从序列k-mers稀疏矩阵中提取特征向量。2020年,Yi等人 [6] 提出了一个堆叠集成计算模型RPI-SE,该模型集成了梯度提升决策树(Gradient Boosting Decision Tree, GBDT)、支持向量机(SVM)、和极端随机树算法,同样基于序列信息来预测LPI。
深度学习同样在长链非编码RNA与蛋白质作用关系预测领域有了很广泛的应用。在2020年,Zhang等人 [7] 提出了一种基于卷积神经网络的并采用了复制填充技巧的深度学习模型,称为LPI-CNNCP。在2021年,Shen等人 [8] 提出了一种基于GNN的非编码RNA-蛋白质相互作用预测方法,称为NPI-GNN,它能够根据网络信息和序列信息预测新的交互。在2021年,Li等人 [9] 提出了一种新的多通道胶囊网络框架,集成用于LPI预测的多模特征,叫做Capsule-LPI。在2021年,Jin等人 [10] 提出一种基于图自动编码器和协同训练的端到端深度学习模型来预测长链非编码RNA和蛋白质相互作用,称为LPIGAC,该方法通过对在长链非编码RNA网和蛋白质网上实现的图自动编码器进行协同训练来提取特征。
此外,许多用于长链非编码RNA-蛋白质关联预测的半监督学习方法都用到了矩阵分析相关知识。Ge等人 [11] 通过两步传播过程在二分网络上以矩阵迭代形式预测lncRNA-蛋白质相互作用。Zhang等人 [12] 则提出了一种线性邻域传播方法来解决这个问题。Zhao等人 [13] 综合了随机游走和邻域正则化逻辑矩阵分解两种算法,来得到潜在的长链非编码RNA-蛋白质相互作用打分矩阵。Zhang等人 [14] 在非负矩阵分解的基础上添加了图正则化来进一步改善模型性能。
在本文中,我们提出了一种利用矩阵补全算法来预测长链非编码RNA-蛋白质相互作用关系的模型,称为LPIMC。我们的模型首先基于长链非编码RNA与蛋白质的相似矩阵及相关性矩阵构建了一个异构网络,然后使用将矩阵补全问题转化为最小化核范数方法来补全目标矩阵,最终得到两者的预测打分矩阵,以从中获取潜在的长链非编码RNA-蛋白质关联。
2. 数据获取
我们的数据集包含从NPInter v2.0数据库 [15] 中下载筛选得到8112条长链非编码RNA-蛋白质关联信息,其中涉及到了3046个长链非编码RNA和136个蛋白质。它们的序列信息已经被NONCODE数据库 [16] 和UniProt数据库 [17] 证实。为了考察模型在不同数据集上的表现,我们还从lncRNome [18] 上采集了一个新数据集,经过筛选去掉和NPInter重复的部分,最终得到了2729对相互作用数据,涉及到1184个长链非编码RNA和9个蛋白质。
2.1. 构建邻接矩阵
我们首先将3046个长链非编码RNA与136个蛋白质进行了重新编号,并由此将此前的8112条关联信息转化为了一个n × m维的邻接矩阵,表示为
,n = 3046表示长链非编码RNA的数量,m = 136表示蛋白质的数量。对应的,若
,则代表第i个长链非编码RNA与第j个蛋白质之间存在关联;若
,则代表第i个长链非编码RNA与第j个蛋白质之间无已知关联信息。
2.2. 构建相似性矩阵
这里采用高斯核(GIP)相似性来 [19] 分别构建长链非编码RNA与蛋白质的相似性矩阵。高斯核相似性矩阵是利用邻接矩阵Y的拓扑信息构建的。在这里,使用
来代表长链非编码RNA的GIP相似性矩阵,使用
来代表蛋白质的GIP相似性矩阵,用长链非编码RNA举例说明,使用AP向量来代表当前RNA与其他所有蛋白质的关联信息,AP向量为从邻接矩阵Y中获得的当前RNA所在的具体行,然后使用下面公式来计算第i个与第j个长链非编码RNA的GIP相似性:
,
其中
是针对于长链非编码RNA的GIP相似性正则化核带宽参数,
是源带宽参数。同理,
的计算方法与之类似。
3. 算法实现
算法思想为将长链非编码RNA-蛋白质关联预测问题转换为矩阵补全问题,然后使用最小化核范数来求解得到预测矩阵。首先,基于上面构建的长链非编码RNA与蛋白质相互作用邻接矩阵Y和两者各自的GIP核相似性矩阵
与
,我们搭建了一个异构的长链非编码RNA-蛋白质网络T视为目标矩阵,表示如下:
不难得出目标矩阵T为
维,构建目标矩阵T的目的是未来使用求解得来的长链非编码RNA-蛋白质预测得分来填充缺失值。由于目标矩阵为低秩的,在此我们将矩阵补全问题转换为最小化目标矩阵秩的问题 [20]。众所周知,最小化目标矩阵秩在计算起来是NP难度的,故通常将秩最小化问题转化为最小化矩阵核范数的问题,该结论已经被研究人员证明 [21]。我们在研究过程中借鉴了ADMM算法 [22],ADMM是一个不仅可以分解,并且在收敛性能上表现优越的算法模型。故该问题可转化为:
,
代表X的核范数,Ω是目标矩阵节点的坐标集,与已知的长链非编码RNA-蛋白质对相对应。
是Ω上的正交投影算子。
除此之外,为了进一步提升ADMM算法,我们在方程中加入了正则化项和矩阵值约束,以确保得到的预测打分落在(0, 1)的范围内,因为(0, 1)范围外的打分没有任何意义。由此得到的关键方程如下:
其中
是表征误差项的参数,
代表X中的所有元素都在(0, 1)的范围内。我们在前面的函数中还引入了一个辅助矩阵W来进一步提高模型收敛性能。最终目标函数如下:
,
在上述公式的基础下,增广拉格朗日函数为:
其中Y是拉格朗日乘数,
代表惩罚参数。我们将W, X和Y初始化为
,然后进行迭代,在第k次迭代时,根据ADMM可以计算得来
、
和
。当迭代终止时,最终的预测矩阵
可以根据之前T的形式进行划分,其中
代表长链非编码RNA与蛋白质的预测关联矩阵,
和
为长链非编码RNA与蛋白质各自的相似性矩阵,此时
中之前存在的空白值被填满,填充值即为长链非编码RNA与蛋白质潜在关联预测打分。预测矩阵及迭代终止条件表示如下:
,
其中默认参数设置为:
;
;
;
;
。模型流程图如图1所示。
4. 性能评估
模型评估方面,我们采用AUC (ROC曲线下面积)作为评估指标。ROC即受试者工作特性曲线,其横坐标为假阳性率(False Positive Rates, FPR),纵坐标为真阳性率(False Positive Rates, TPR),计算公式为:
,
。对应的值由TP,FP,TN,FN计算而来。其中TP为真实为正类且预测同为正类的样本,FP为真实为负类而预测为正类的样本。同理,TN为真实为负类且预测同为负类的样本,FN为真实为正类却被预测为负类的样本。
我们在整理筛选后的数据集上进行了10次5折交叉验证,在5折交叉验证中,8112个确认的长链非编码RNA-蛋白质关联被随机分为5组,每个集合被依次当作测试集,其他四个集合被合并为训练集,并使用在训练集上训练出的模型来预测测试集中的关联得分,最后通过计算得到模型的平均AUC值为0.98 ± 0.01。为了证明模型的泛化能力,我们还额外从lncRNome数据库上采集了一个新数据集,经过筛选得到了2729对相互作用数据,涉及到1184个长链非编码RNA和9个蛋白质。经过LPIMC模型训练测试得到5折交叉验证平均AUC为0.985 ± 0.003,模型表现性能较在NPInter v2.0上表现更为出色,据分析可能是因为在该数据库中已知长链非编码RNA与蛋白质关联占比更高所致。模型在两个数据集上的ROC曲线图如图2所示。
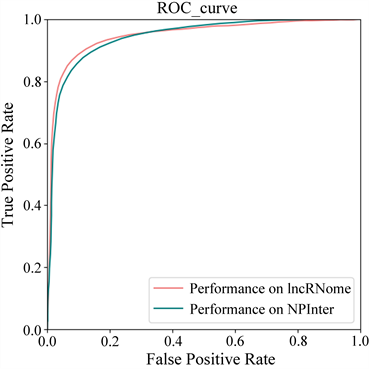
Figure 2. The ROC curve on two dataset
图2. 两个数据集上的ROC曲线
5. 结论
本文中,我们提出了一种利用矩阵补全策略来进行长链非编码RNA与蛋白质相互作用关系预测的模型(LPIMC)来预测两者的潜在关联。实验数据表明,尽管所能利用的关联数据十分有限,模型仍取得了优良结果,且不过分依赖于长链非编码RNA与蛋白质本身的特殊特性,这些都表明了LPIMC模型可以扩展到类似的分类任务。长链非编码RNA与蛋白质的关联预测数据集属于较大规模数量级的数据集,而由于LPIMC使用了较少的计算资源,故它的时间效率也表现出色。同时,我们也对LPIMC模型的改良有了一些设想,例如将矩阵补全算法和当前大热的深度学习及图学习相结合,亦或在数据集层面下功夫,获取更高质量且更均衡的数据集。这些都能帮助提升模型预测性能。