摘要: 目的:探讨儿童以视神经炎(ON)为首发症状的抗髓鞘少突胶质细胞糖蛋白(MOG)相关疾病同时合并抗N-甲基-D-天冬氨酸受体脑炎(NMDAR)的临床特点。方法:回顾分析作者医院儿内科收治的1例以视神经炎(ON)为首发症状的抗髓鞘少突胶质细胞糖蛋白(MOG)相关疾病合并抗N-甲基-D-天冬氨酸受体脑炎(NMDAR)患儿的临床资料。结果:该女性患儿,9岁,因头痛、视力下降起病,在其脑脊液检查中发现抗NMDAR (+),血清MOG (+),颅脑核磁提示双侧半卵圆中心少许脱髓鞘病变;双侧视神经异常改变,诊断为视神经炎、MOG抗体病合并抗NMDAR脑炎重叠综合征。在给予丙种球蛋白及大剂量激素冲击治疗后,患儿头痛缓解,视神经损伤情况得到控制。结论:MOG抗体病合并抗NMDAR脑炎较单一抗体发病的临床特征不典型,但预后相对较差,临床上尽早完善相关抗体检测有助于疾病的诊断,尽快实行有效的免疫治疗可使患儿受益。
Abstract:
Objective: To investigate the clinical features of anti-myelin oligodendrocyte glycoprotein (MOG)- related diseases with optic neuritis (ON) as the first symptom in children combined with anti-N- methyl-D-aspartate receptor encephalitis (NMDAR). Methods: The clinical data of a child with an-ti-myelin oligodendrocyte glycoprotein (MOG) related disease and anti-N-methyl-D-aspartate re-ceptor encephalitis (NMDAR) with optic neuritis (ON) as the first symptom admitted to the pediatric Department of our hospital were retrospectively analyzed. Results: The 9-year-old female child presented with headache and decreased visual acuity. In her cerebrospinal fluid examination, an-ti-NMDAR (+) and serum MOG (+) were found. Brain MRI showed a little demyelination in the center of bilateral semiovale. Lesions; abnormal changes in bilateral optic nerves, diagnosis of optic neuri-tis, MOG antibody disease combined with anti-NMDAR encephalitis overlap syndrome. After giving gamma globulin and high-dose steroid pulse therapy, the child’s headache was relieved, and the optic nerve damage was controlled. Conclusion: The clinical features of MOG antibody disease com-bined with anti-NMDAR encephalitis are less typical than that of single antibody, but the prognosis is relatively poor. Improving the detection of relevant antibodies as soon as possible is helpful for the diagnosis of the disease, and implementing effective immunotherapy as soon as possible can make the children benefit.
1. 前言
髓鞘少突胶质细胞糖蛋白(myelin oligodendrocyte glycoprotein, MOG)是免疫球蛋白超家族的一员,是一种表达于外髓鞘上的跨膜蛋白,具有一定的免疫原性,可被自身抗体识别,从而引起免疫介导的中枢神经系统脱髓鞘疾病 [1]。MOG-IgG阳性患者临床表现多样且复杂,在成人患者中以视神经脊髓炎谱系病(NMOSD)和横贯性脊髓炎(LETM)多见,而在儿童患者中主要表现为急性播散性脑脊髓炎(ADEM)、复发性炎症性视神经炎(RION)。据文献报道,MOG相关疾病是儿童视神经炎(ON)最常见的原因,占儿童急性脱髓鞘事件的50% [2]。据Moon Yeji等报道,ON初次发病的平均年龄约在5~10岁,其中女性患儿居多。多数患儿初次发病即以双侧视神经炎表现,少数患儿可演变成为复发性视神经炎。在视神经炎急性期,主要表现为视盘肿胀,少部分可表现为乳头周围出血;在慢性期,绝大多数可出现视神经萎缩 [3]。
抗N-甲基-D-天冬氨酸受体(NMDAR)脑炎与脑脊液中针对NMDAR的GluN1亚基的IgG抗体有关,通常好发于女性青年患者,临床上以精神异常和行为障碍为主要表现,如癫痫发作、运动、记忆及言语障碍、意识水平下降、自主神经功能障碍和中枢通气不足为特征表现 [4]。
同时存在MOG脱髓鞘疾病和抗NMDAR脑炎的概率较低,可能导致自身免疫性疾病的多样性,其机制尚不清楚。有文献指出,两种及两种以上抗体共存可能是由自身免疫性疾病的缺陷引起的。
2. 病情介绍
女性患儿,9岁,因“头痛半月余,加重伴视力下降2天”于2021-09-13入住山东大学第二医院儿科。患儿半月余前无明显诱因诉阵发性头痛,以头颅颞部及顶部为著,多于午后及夜间入睡前出现,晨轻暮重,发作频率约为3次/周,无发热,无头晕目眩,无视物模糊,无恶心、呕吐,无惊厥,无情绪改变,院外未予规范化诊治,头痛可自行缓解。入院前2天,患儿突然出现双眼视物模糊不清,视力下降,右眼较著,甚至出现黑朦,左眼视力逐渐加重至10 cm可见度。偶诉眼部疼痛,无发红、流泪及分泌物。患儿头痛也较前频繁,无发热,无肢体无力及麻木,无运动障碍。入院1周前患儿曾有呼吸道感染病史,病初伴有发热情况,自行口服抗菌药物治疗后体温恢复正常。入院时体格检查结果如下:体温37.3℃,脉搏86次/分钟,呼吸频率22次/分钟,血压117/71 mmHg,体重43.5 kg神志意识清楚,精神反应一般,近、远期记忆力无明显下降,无明显语音障碍。眼睑无浮肿,结膜无充血。鼻唇皱褶和牙齿对称,眼球活动自如,双侧瞳孔3 mm,等大等圆,对光反射迟钝,右眼不能视物,左眼可视距离不足10 cm。心肺腹部查体未见明显异常。颜面部痛触觉,味觉,听觉,四肢肌张力和深浅感觉均未见异常;病理反射未引出;脑膜刺激征阴性。入院当天完善眼底检查示双侧视乳头水肿;视力:左侧0.1,右侧0.02;完善视觉诱发电位示双侧P100潜伏期延长,右侧波幅降低。颅脑核磁 + DWI示:双侧半卵圆中心见少许斑点状长T1稍长T2信号灶,T2-FLAIR呈高信号,DWI未见明显高信号,脑液腔未见明显异常,中线结构居中。扫及双侧视神经增粗、扭曲,边緣欠规则,信号不均匀,视神经鞘轻度增厚。双侧半卵圆中心少许脱髓鞘病变;双侧视神经异常改变,结合临床,考虑视神经炎(见图1)。动态脑电图提示为异常儿童脑电图,背景基本节律偏慢双侧前头部不规则尖形慢波不同步发放,右侧著,清醒期著。入院后第二天对患儿进行腰椎穿刺,进行脑脊液分析,脑脊液常规、生化及病毒分析结果未见明显异常。同时我们完善了血清及脑脊液自身免疫性抗体、寡克隆抗体及脱髓鞘抗体等,结果显示,患儿脑脊液中存在抗NMDAR抗体1:32 (+),血清中存在中枢神经系统脱髓鞘疾病抗体:抗MOG抗体IgG 1:32阳性(见图2(A)~(C))。结合患儿病情及检查结果,该患儿被诊断为“视神经炎;MOG抗体病合并抗NMDAR脑炎重叠综合征”成立。我们给予丙种球蛋白封闭及甲泼尼龙的冲击治疗。第一疗程激素冲击完成后,患者的头痛明显减轻,视力逐渐恢复。请眼科会诊提示:双眼RAPD (+),ZOP:17 mmHg 19 mmHg;V: R: 0.04 L: 0.5;视野:右视野鼻上颞下缺失,余视敏度下降,左上方视野部分缺失;建议继续激素冲击治疗。复查视频脑电图提示了异常儿童脑电图,双侧前头部尖形慢波不同步发放,右侧著,睡眠期著。在激素冲击第11天,她复查颅脑核磁 + DWI提示右额叶脑白质内见少许斑点状长T1稍长T2信号灶,T2-FLAIR呈高信号,DWI未见明显高信号,脑液腔未见明显异常,中线结构居中。扫及部分鼻窦粘膜略厚。双侧视神经略粗,局部略扭曲,边缘欠规则,信号不均匀,压水像呈高信号,双侧眼球形态可,未见明显异常信号,眼肌走行自然,未见明显异常,眶内脂肪间院清晰。右额叶少许异常信号;双侧视神经异常改变。在随后的治疗中,该儿童未再诉头痛,视力日渐好转,我们逐渐减少了甲泼尼龙的给药量直至停药。3个月后,该患儿复查血清中枢神经系统脱髓鞘-MOG抗体,提示1:10,已经恢复至起始浓度梯度(见图2(D))。在后续随访中,该患儿视力虽然未达到生病前的视力水平,但一直在逐渐恢复,在最近的随访中患儿左眼视力已接近1.0,右眼视力相差较大,仅在0.4左右(患者入院时、治疗后及随访期间的情况见表1)。

Figure 1. AB: optic nerve thickening and twisting changes; CD: there are a few spot-like abnormal changes in the right white matter
图1. AB:视神经增粗、扭曲改变;CD:右侧脑白质内见少许斑点状异常改变
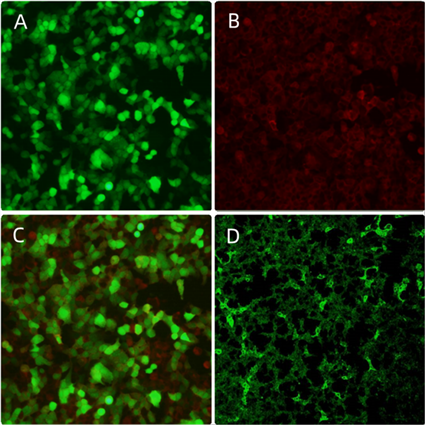
Figure 2. A: serum MOG protein; B: positive anti-MOG antibody; C: antibody superposition effect; D: negative result of serum MOG antibody in later stage
图2. A:血清MOG蛋白;B:血清抗MOG抗体阳性;C:抗体叠加效果;D:患儿后期复查血清MOG抗体阴性结果

Table 1. Summarizes the clinical situation, treatment and examination results of the patient
表1. 总结该患者的临床情况、治疗和检查结果
3. 讨论
近年来,MOG-IgG在中枢神经系统炎性脱髓鞘疾病患者血清中的作用已逐渐被认识到。MOG是中枢神经系统(CNS)髓鞘的主要成分之一,虽然MOG占中枢神经系统成分的不到0.5%,但其许多表位在啮齿动物和人类中具有高度的免疫原性。视神经炎(ON)是指视神经的炎性脱髓鞘病变,可导致急性或亚急性视力丧失,是青年人视力受损、甚至失明的主要原因。MOG-ON抗体病的治疗分为急性期和缓解期。急性期治疗的目的是最大限度地挽救视觉功能,防止或减少和延迟对神经系统的进一步损害,多采用大剂量激素冲击治疗。缓解期主要是为了降低复发率以及失明和残疾的严重程度,通常根据患者复发风险来选择是否采用长期免疫调节治疗。我们可以通过血清学检查判断复发风险,如患者血清中MOG-IgG持续阳性,这提示患儿很大可能会复发视神经炎;相反,若MOG抗体滴度逐渐降低,则提示疾病处于恢复期,复发风险相对较小。在本案例中,患儿诊断明确后即予丙种球蛋白及大剂量糖皮质激素冲击治疗,经过一定疗程的治疗,患儿神经损伤未再加重,继予续贯减量治疗,出院时患儿双眼视力得到明显恢复。
抗NMDAR脑炎是机体对NMDAR NR1亚基产生特异性反应的一种可治性、免疫相关性脑炎,是儿童最常见的自身免疫性脑炎(autoimmune encephalitis, AE),占儿童抗体阳性自身免疫性脑炎的90%以上 [5]。从性别及年龄分布来看,青少年女性多见;从发病季节来看,4~9月份多发;患儿常以精神异常、意识水平下降及不同程度的行为障碍,如烦躁不安、失眠、运动失调、胡言乱语及记忆偏差等。对于抗NMDAR脑炎治疗的一线用药有糖皮质激素、血浆置换和丙种球蛋白。2021年国际儿童抗NMDAR脑炎治疗的共识中提出,对于轻症患儿推荐静脉使用甲泼尼龙20~30 mg/(kg·d),最大1000 mg/d,疗程3~5 d;对于重症患儿推荐联合应用免疫球蛋白和(或)血浆置换作为一线治疗。对免疫治疗反应好的患儿推荐3~6个月的免疫维持治疗,对反应一般的患儿推荐6~12个月的免疫维持治疗,对免疫治疗反应差的患儿推荐1~2年的免疫维持治疗 [6]。由于部分患儿康复后抗NMDAR抗体仍可长期存续,所以没有明确证据表明抗体滴度与疾病的进展及演变有明显的关联性。在本例报道中,患儿短期内视神经受损进行性加重,幸运的是对糖皮质激素治疗较敏感,急性期过后,又给予3个月的免疫维持治疗。抗N-甲基-D-天冬氨酸受体(NMDAR)脑炎的复发率很高,据统计,约10%~20%的患者在发病后2年内复发。Yamada等报道了一名13岁的左撇子女孩,因出现意识水平低下和双侧强直阵挛发作就诊,根据脑脊液(CSF)等检查提示诊断为抗NMDAR脑炎,给予静脉大剂量甲泼尼龙冲击和免疫球蛋白的治疗,患儿症状得到了明显缓解。但在其23岁时,她突然出现轻度右侧肢体麻木、构音障碍、失乐症和强直阵挛性癫痫发作 [7]。这刷新了抗NMDAR脑炎的最长复发周期,也给我们敲醒了警钟,对于这类患儿要做好长期随访的准备。
据文献记载,少数抗NMDAR脑炎可以与MOG抗体阳性脱髓鞘性疾病重叠发病,发病年龄多倾向于儿童,成年人较少见,我们称之为重叠综合征(MNOSD)。据文献报道,抗NMDAR脑炎合并MOG抗体病时发病症状相对单纯抗NMDAR脑炎较轻 [8],常以精神行为异常、认知功能障碍为主;但重叠综合征的患儿预后较差 [9]。在本例报道中,患儿虽然患有重叠综合征,但症状仅有脱髓鞘疾病的相关表现,如双侧视神经的累及,而精神行为及认知功能没有受到明显损害,这也进一步印证了上述说法。
综上所述,当在临床中遇到疑似自身免疫性脑炎或者脱髓鞘相关疾病的患儿时,早期完善血清学、脑脊液及影像学检查是明确诊断的重要前提。不能仅仅依靠临床症状去预判病情的严重程度及疾病的转归。明确诊断后尽早予以大剂量糖皮质激素等一线免疫治疗对患儿病情的演变及预后有重要的价值。当然,做好长期且细致的随访对我们了解此类疾病的转归有着莫大的帮助。