1. 引言
近几年来,随着基因组学和蛋白质组学的不断发展以及医学前沿技术的迅速突破,免疫疗法作为多种癌症的治疗方法不但获得了巨大的成功,并且具有良好的应用前景。免疫疗法是一种利用人体自身免疫系统来对抗癌症的方法,对癌症治疗领域的发展至关重要。随着全球免疫学、医学和细胞生物学等相关领域的科学研究工作不断进步,全球免疫疗法的研究进展也正在快速提升,并且已经取得很多研究成果。继手术、化疗和放疗等传统治疗方法后,免疫疗法技术已经逐渐成为了治疗癌症领域的新兴方法。目前,发展较为迅速的免疫疗法主要包括TCR-T (T Cell Receptor-Gene Engineered T Cells)和CAR-T (Chimeric Antigen Receptor T Cells)等疗法 [1]。CAR-T疗法对血液肿瘤疾病取得良好的治疗效果,然而CAR-T免疫疗法存在着一些缺点。首先是它对肿瘤细胞的识别机制,CAR-T是通过基因编辑技术人工设计的单链抗体片段(CAR),该片段对识别癌细胞膜表面表达的抗原较为有效,然而癌细胞内部还存在着大量的实体瘤抗原,CAR-T细胞并不能对胞内抗原进行识别。因此,CAR-T疗法对实体肿瘤的治疗效果并不十分理想。然而,通过TCR-T疗法设计的带有编辑过TCR的T细胞不但可以识别癌细胞表面的抗原,而且还能识别MHC递呈的癌细胞内部抗原。TCR-T细胞上的TCR通过结合MHC分子递呈出来的抗原肽段识别癌细胞,进而主动攻击将癌细胞杀死,达到治疗癌症的目的。
2. 现有研究技术
2.1. TCR和肽的相互作用原理
TCR-T细胞是通过自身表面的TCR识别MHC分子递呈的抗原肽而产生相互作用的,TCR-T细胞上的TCR与MHC I类分子呈递的肽结合的过程如图1所示 [2]。抗原呈递细胞(APC)内的抗原蛋白质分子首先被蛋白质裂解酶水解为大量的多肽。接着,多肽通过抗原加工相关的转运蛋白的转运,输送到内质网中,MHC I类分子与内质网上的多肽结合后,呈递到细胞膜表面。最后,TCR识别MHCⅠ分子呈递的多肽,两者结合之后产生胞间信号,T细胞和肿瘤细胞之间出现免疫应答反应,进而杀死癌细胞,达到治疗肿瘤的目的。然而,并不是所有的MHC分子所呈现的多肽都能和TCR发生相互作用,其中,很多多肽片段不具有免疫原性,也就不会被TCR所识别。因此,如何从大量非结合TCR中筛选出特异性识别肿瘤抗原肽的TCR,成为了目前TCR-T疗法中一大挑战。通过高通量设备和实验方法,从大量的TCR和候选肽中筛选出可以相互作用的配对序列显然是不现实的。这样做不仅耗费大量的时间,而且需要很多资金支持。如何快速准确地预测TCR和肽之间的相互作用已经成为近些年来生物医学领域重点研究方向。
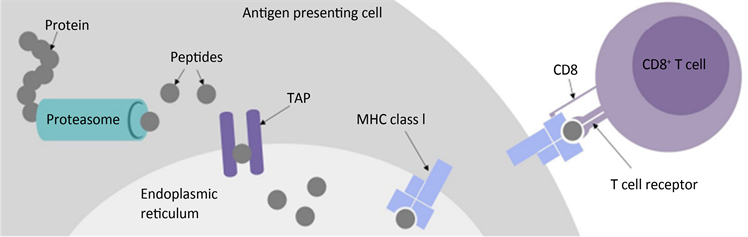
Figure 1. The process of the TCR and peptide interaction
图1. TCR和肽的相互作用的过程
2.2. 现有研究方法
随着人工智能技术的不断发展,生物医学与人工智能的结合逐渐成为新的突破点。人工智能领域的机器学习和深度学习技术作为一种强有力的方法为预测TCR和肽之间相互作用提供了指导。到目前为止,现有的预测TCR-肽的相互作用的方法主要基于分子动力学、机器学习和深度学习三类。在预测TCR和肽结合的亲和力的早期研究当中,Michielin等人利用热力学积分法计算自由能,基于分子动力学分析结合的TCR和肽之间的自由能的差异,来反映TCR和肽之间结合亲和力的强弱 [3]。该方法对于预测TCR和肽亲和力具有重大的生物意义。2019年,Gielis等人研发了一种基于机器学习的随机森林分类模型TCRex预测TCR和肽的相互作用 [4]。该模型从McPAS-TCR数据库和VDJdb数据库中筛选出TCR的β链序列和配对的肽序列,训练和评估TCRex模型。该分类器使用5倍分层交叉验证方法计算了每个表位特异性模型的ROC曲线下面积(AUC)值。模型的AUC至少为0.7,体现出该模型较强的泛化能力。Jurtz等人在深度学习的基础上设计了一种基于卷积神经网络的分类模型NetTCR [5],该模型能够预测TCR和MHC I等位基因HLA-A*02:01呈现的肽之间的相互作用。NetTCR模型依赖于肽的氨基酸序列和TCRβ链的CDR3区域作为输入,这两个序列都是使用BLOSUM50矩阵进行特征表示。CNN非常适合处理长度不同的未对齐肽和TCR序列,使用卷积滤波器扫描两个输入,CNN能够整合整个输入序列中不同过滤器发现的模式的信息。接着对模型进行训练和评估。结果表明,该模型可以从大量非结合TCR中识别结合给定同源MHC呈递的肽靶点的TCR。
然而这些现有的研究方法虽然能够对TCR和肽的相互作用进行预测,但是由于TCR-肽序列配对的多样性和可用训练数据有限,这些模型对于已经训练和测试过的数据预测效果较好,然而对未覆盖的或潜在的肽或TCR序列的新数据预测能力一般,模型的泛化能力和稳定性较差 [6]。因此,需要寻找一个较好的特征工程方案对TCR和肽序列数据进行特征编码,并且开发出一种基于深度学习的高性能和高稳定性的方法,这对预测TCR和肽的相互作用至关重要 [7]。
3. 基于深度学习的TCR和肽的相互作用预测模型
基于上述预测TCR和肽的相互作用模型的研究现状和存在的问题,本文从两个不同角度分别对TCR和肽进行数值化表征,提出了一种基于深度学习的神经网络模型(C-L-M)预测TCR和肽的相互作用。C-L-M模型从不同的数据挖掘角度出发,不仅为TCR和肽的相互作用预测提供了新的认知,而且为TCR-T免疫治疗方法中TCR和肽的初步筛选提供了一定的指导作用。
3.1. 实验数据集
本实验所有的数据都是从TCRdb数据库中经过一定条件筛选出来的,一共包含3416对TCR和肽相互配对的阳性数据和3416对的阴性数据,一共包含6832对数据,这些数据都是经过实验验证的。数据集按7:2:1划分为训练集、验证集和测试集。本研究所用数据集的分布如表1所示。

Table 1. Distribution of the datasets used in this paper
表1. 本文所用数据集的分布
3.2. 数据的预处理和特征表示
对TCR和肽的氨基酸序列构建一个有效的特征数值化方法,将会对预测两者相互作用结果的准确率和精准度造成直接影响。本研究首先对人类的二十种氨基酸用自定义的getDict函数定义了一个字典,把TCR和肽的氨基酸序列作为一个整体(TCR-肽序列),使用自定义的toToken函数把字符序列转换为数值序列。接着从不同角度对TCR-肽数值序列进行特征提取。一个角度对TCR-肽数值序列使用One-Hot编码方式表示 [8],另一个角度是对TCR-肽数值序列用Embedding编码方式来表示 [9]。通过对TCR-肽序列从两种从不同角度分别进行特征提取,把这两种表示方法相结合,对TCR和肽的相互作用预测进行特征表示。
3.3. C-L-M模型的构建
由于一维卷积神经网络(Conv1D)和循环神经网络(LSTM)在处理文本序列具有很好的性能,而多层感知机(MLP)具有处理高维、复杂的数据的能力。因此,我们把Conv1D、LSTM和MLP进行了一个组合,设计了一种基于深度学习的模型C-L-M。让Conv1D去处理One-Hot编码方式表示的TCR-肽序列的特征矩阵,让LSTM去处理用Embedding编码方式来表示的TCR-肽序列的特征矩阵。最后,用Concatenate()函数把两个输出向量的特征进行融合,输入到设计的MLP神经网络中,对TCR和肽的相互作用进行预测。基于深度学习的预测TCR和肽的相互作用的C-L-M算法流程图如图2所示。
3.4. 模型的训练和超参数设置
本研究把One-Hot编码和Embedding编码TCR-肽序列分别经过Conv1D和LSTM进行处理。Conv1D神经网络是由2个卷积层、2个池化层和1个全连接层构成,卷积核数量都是64,全连接神经元数量为128。LSTM神经网络是由2个LSTM和一个全连接层构成,神经元个数分别为,64、64和128。随后,把两个输出向量用Concatenate()函数进行特征融合,输入到具有3个全连接层的MLP神经网络中,神经
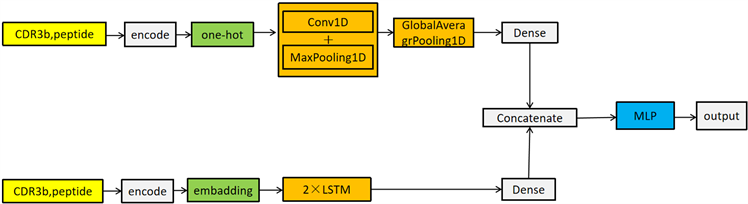
Figure 2. Flow chart of the C-L-M algorithm based on deep learning
图2. 基于深度学习的C-L-M算法流程图
元个数分别为128、64,和32。Conv1D、LSTM和MLP中的隐藏层全部采用采用ReLU函数作为激活函数。在全连接层之后加入Dropout层,rate设置为0.2。加入Dropout层的作用是防止网络过拟合,随机让一定数量的神经元停止工作,这样可以提高网络的泛化能力 [10]。最后,加一个全连接层作为输出层,神经元个数为1,Sigmoid作为激活函数(表达式如式(2)所示),输出结果。本研究使用二值交叉熵(Binary_crossentropy)作为C-L-M模型的损失函数(表达式如式(3)所示),损失函数的目的是为了计算TCR和肽的相互作用的概率值和实际值之间的误差,其中,
为真实值,
为预测值。本研究使用的优化器是RMSprop算法(Root Mean Square prop),学习率为0.001。使用RMSProp算法对神经网络中神经元的权重W和偏置b的梯度进行更新 [11]。其中,ReLU、Sigmoid和Binary_crossentropy的公式如下:
(1)
(2)
(3)
4. 实验结果与分析
4.1. 实验环境
在软件环境方面,操作系统为Windows 10,Python环境版本为Python 3.9,使用的框架版本是Keras 2.5和Tensorflow 2.5。在硬件环境方面,使用的CPU是AMD Ryzen 7 4800U with Radeon Graphics 1.80 GHz,计算机的内存是16G,使用的显卡是GTX 1060。
4.2. 性能指标
把预测TCR和肽的相互作用的模型构建完成后,我们需要对模型的性能进行一个评估。不同种类的模型具有不同的评估指标,这些指标能够客观的评估模型性能的好坏。选择合适的指标可以帮助我们对模型的性能进行调整。通过把不同的指标进行比较,从各种模型中选择最优的一种作为最终模型。本研究是对TCR和肽的相互作用的预测结果进行一个分类,因此,采用分类性能的评价指标作为评估模型的标准。本研究选择了ROC曲线、Precision、Recall和AUC作为C-L-M模型评估的指标。其中,TP代表真阳性,即正确识别TCR与配对肽结合;TN代表真阴性,即正确识别TCR不结合配对肽;FN代表假阴性,错误识别TCR不结合配对肽。Precision和Recall的公式如下:
(4)
(5)
4.3. 实验结果和分析
为了评估C-L-M模型的性能,我们将C-L-M模型与基于机器学习的随机森林分类模型和Jurtz等人提出的基于卷积神经网络的NetTCR分类模型在本文研究的数据上进行对比。C-L-M模型与其他两个模型的ROC曲线对比如图3所示。由此图可以看出,C-L-M模型的ROC曲线相对与随机森林和NetTCR更加靠近左上角,这表明C-L-M模型要比其他两个的预测效果都要好。而C-L-M模型与其他模型在其他指标的对比结果如表2所示,从该表我们得知,C-L-M模型的AUC为0.8513,而随机森林分类模型和NetTCR分类模型在AUC方面表现一般,分别为0.8263和0.8039。与随机森林和NetTCR模型相比,C-L-M模型在AUC方面分别提高了0.025和0.0474。在其他指标方面,与NetTCR相比,C-L-M模型的Precision提升了0.0489。与随机森林相比,C-L-M模型的Recall提高了0.0761。因此,C-L-M模型在预测TCR和肽的相互作用的性能有着不错的表现。

Figure 3. ROC curve comparison of C-L-M and other classification models
图3. C-L-M与其他分类模型的ROC曲线对比图
Table 2. Comparison of various indexes of C-L-M and other classification models
表2. C-L-M与其他分类模型的各种指标对比
5. 结论
深度学习形式的人工智能和免疫生物学的结合对于进一步了解TCR和肽的相互作用至关重要。为了从大量TCR和肽的氨基酸序列中快速地、高通量地筛选出具有高亲和力和稳定性的TCR-肽对序列,本文提出了一种基于深度学习的分类模型C-L-M,该模型从两个不同的角度对TCR-肽序列进行特征提取,并把Conv1D、LSTM和MLP神经网络进行组合,实现TCR和肽的相互作用预测。实验结果显示,与其他分类模型相比,C-L-M模型在特征提取方面和处理复杂数据方面性能表现更好。该方法不仅提高了TCR-T疗法中TCR和肽的筛选效率,而且对细胞免疫治疗方法的研究起到了推动的作用。