1. 引言
化石燃料的消耗逐年增加导致二氧化碳(CO2)在大气中不断累积,造成了严重的温室效应和海洋酸化等环境问题,迫使人们寻找有效途径来降低大气中CO2的浓度 [1] [2] [3] 。近年来,已经有多种高效的CO2转化技术开发出来,包括生物催化、光催化和电催化方法等。其中,电催化CO2还原反应(CO2RR)生成甲烷(CH4)作为一种有前途的途径受到了广泛关注 [4] [5] 。然而,由于二氧化碳具有极佳的热力学稳定性和较高的活化能垒使得该反应较难发生 [6] 。另外,析氢反应(HER)因其较低的过电位而成为CO2RR的竞争副反应。因此,设计低过电位且能抑制HER的新型CO2RR电催化剂具有重要意义 [7] [8] 。
近年来,由于纳米技术的发展,人们可以在纳米尺度进行结构微调从而控制电催化剂的结构,使其拥有更好的稳定性、催化活性和产物选择性 [9] [10] 。例如,活性位点高度分散的原子层厚度级别的单原子催化剂(SACs)、双原子催化剂(DACs)和三原子催化剂(TACs)等,因其具有过电位低、转化效率高、产物选择性好以及抑制CO2RR过程中HER的能力,所以具有良好的应用前景 [11] [12] 。具有Mn、Mo、Ru和Ti等过渡金属原子的SACs和DACs已被多篇报道证明具有较好的CO2RR催化活性 [13] [14] [15] [16] [17] 。最近,根据一些报导,TACs因其较高的金属原子负载率、较高的金属原子利用率和较大的接触面积有望成为性能更好的CO2RR催化剂 [18] [19] [20] 。值得注意的是,TACs由金属三聚体和支撑材料组成,其合成工艺复杂且稳定性较差,因而对支撑材料有较高要求。合适的支撑材料可以防止过渡金属三聚体的聚集,从而降低合成难度,并且它们之间强烈的界面相互作用可以提高催化剂的稳定性 [20] [21] 。多孔氮化石墨烯(C2N)是一种存在周期性均匀空腔的二维材料,是氮掺杂石墨烯的一种衍生物 [22] 。其空腔周围环绕着6个氮原子(N6空腔),被认为是金属三聚体的理想载体。其本身较高的热稳定性有利于合成稳定的TACs [23] 。因此,C2N在充当三原子催化剂的支撑材料上有巨大潜力。
因此,在本工作中,通过第一性原理,建立了C2N负载的三原子催化剂3TM-C2N (TM = Mn, Mo, Ru, Ti)模型,并系统地研究了3TM-C2N催化CO2RR生成CH4的性能。首先,计算了3TM-C2N的稳定性及电子结构;其次,从d带中心理论及电子结构的角度,深入分析了3TM-C2N对CO2的吸附与活化;最后,阐明了CO2在3TM-C2N上还原为CH4的最有利路径及其极限电势。计算结果表明,3TM-C2N催化剂结构稳定,能有效吸附和活化CO2,并且对HER有良好的抑制性。其中,3Mn-C2N表现出最好的催化活性,其极限电势为−0.44 V。这些发现不仅为实验上调控C2N基催化剂提供了理论依据,还对开发其他高效的CO2RR电催化剂有一定的指导意义。
2. 研究方法
2.1. 计算参数
基于具有周期性边界条件的密度泛函理论(DFT),本工作中的所有任务都是在Vienna Ab-initio Simulation Package (VASP)中进行计算的 [24] [25] 。计算过程中,将C-2s22p2、N-2s22p3、O-2s22p4、H-1s1以及Mn-3d64s1、Mo-4d55s1、Ru-4d75s1、Ti-3d34s1电子轨道作为价电子,通过投影缀加平面波(Projector Augmented Wave, PAW)方法描述价电子与芯电子间的相互作用,通过广义梯度近似(Generalized Gradient Approximation, GGA)中的Perdew-Burke-Ernzerhof (PBE)方法描述交换关联函 [26] [27] 。为了使总能量的结果更加精确,计算中打开了自旋极化并施加了DFT-D3修正。其中,动能截断设置为450 eV,能量和力的收敛标准分别为10−4 eV和0.05 eV/Å。高斯方法将温度展宽设置为0.01 eV,用于改善电子收敛步中接近费米能级的电子状态的收敛性。结构优化时K空间以伽马为中心(Gamma-centered)进行3 × 3 × 1的网格剖分。在计算的过程中,对于结构中的全部原子进行驰豫。
2.2. 计算公式
本文采用了计算氢电极模型(CHE)计算了标准条件下质子–电子对与氢气的电化学势。
(1)
依据该方法,每个基本反应的吉布斯自由能变化值计算公式如下 [28] :
(2)
其中,
为DFT计算得到的能量差值,
为零点能的变化,T为温度(298.15 K),
为熵的变化。
吸附能大小可以判断CO2吸附在催化剂表面时的稳定程度。吸附能公式如下 [28] :
(3)
其中,
为总能量,
和
分别代表了与吸附分子作用的表面模型和真空中独立的的吸附分子的能量。通常来说,吸附能为负值,表明吸附过程是放热反应,吸附的系统是稳定的。
CO2吸附在催化剂表面的机理可以由d带中心理论阐述,计算d带中心的公式如下:
(4)
其中,
为对应的d带上电子的密度,
为能量。
另外,极限电势(
)是使每个基本步骤都能放热的最小负电位。公式为:
(5)
其中,
为整个CO2RR路径中自由能变化的最大值,e为电子电量 [29] 。
3. 结果与讨论
3.1. 3TM-C2N模型的构建及稳定性分析
优化后的二维C2N晶格参数为a = b = 8.32 Å,与文献中8.30 Å非常接近,说明计算参数设置的比较合理 [30] 。三聚体模型采用了C2N晶格的2 × 2超晶胞,模型中共有48个C原子和24个N原子,如图1(a)所示。在c方向我们设置了15 Å的真空层来避免相邻晶格间的影响。过渡金属三聚体被放置在N6空腔里,每个过渡金属原子都与两个N原子成键,组成三聚体构型。图1(b)~图1(e)显示,Mn、Mo、Ti原子都高于C2N平面。而金属Ru只有两个原子高于C2N平面,一个原子与C2N处于同一平面。

Figure 1. (a) The stable structure of the optimized 2 × 2 C2N supercell; (b)~(e) The stable structure of the optimized 3TM-C2N
图1. (a) 优化后2 × 2 C2N超晶胞的稳定结构图;(b)~(e) 优化后3TM-C2N的稳定结构图
高稳定性是催化剂应用的先决条件,所以我们用内聚能对3TM-C2N催化剂的稳定性进行了评估。内聚能越高,对应的结构越稳定。3TM-C2N催化剂的内聚能为3Mn-C2N (6.67 eV/atom) < 3Mo-C2N (6.71 eV/atom) < 3Ti-C2N (6.76 eV/atom) < 3Ru-C2N (6.79 eV/atom)。它们的内聚能虽然比初始C2N (6.82 eV/atom)略小,但是要高于碳磷化物(4.12~6.45 eV/atom) [31] 和硅烯(3.71 eV/atom) [32] ,说明其具有较高的稳定性。
为了深入理解金属三聚体和C2N之间的相互作用,我们计算了3TM-C2N电子态密度和差分电荷密度,如图2所示。图2(a)~图2(d)电子态密度图显示,在费米能级附近,金属三聚体的d轨道(图中黄色部分)与C2N的中N的p轨道(图中青色部分)之间有明显的重叠峰(图中绿色部分),说明3TM-C2N上的载流子浓度较高,且金属三聚体与C2N的相互作用强烈。尤其在3Ru-C2N中,重叠峰更多,这说明Ru原子与N原子之间相互作用比其他金属原子和N原子之间相互作用更强烈,这是3Ru-C2N催化剂稳定性最高的原因。同时,也正是因为较强的相互作用,导致Ru原子与N原子之间键能更强,键长更短,与图1中有一个Ru原子与C2N处于同一平面的构型相符。
图2(e)~图2(h)差分电荷密度图显示,正电荷在金属原子周围积累(图中青色部分),在N原子附近减少(图中黄色部分),说明电子从金属原子转移到了N原子上,从而使N原子与TM原子结合。

Figure 2. (a)~(d) Density of states (DOS) of 3TM-C2N; (e)~(h) Charge density differences of 3TM-C2N
图2. (a)~(d) 3TM-C2N的电子态密度图;(e)~(h) 3TM-C2N的差分电荷密度图
3.2. CO2在3TM-C2N上的吸附和初始活化
CO2吸附是CO2RR的第一步,也是至关重要的一步。稳定的CO2吸附是CO2RR能继续下去的先决条件。CO2的活化程度一定程度上反映了催化剂的催化性能。如图3所示,CO2能稳定吸附在催化剂表面,并且CO2分子构型发生了明显的变化说明CO2得到了初始活化。为了定量分析,将CO2吸附能和CO2键长键角等信息在表1中列出。CO2吸附在3Mn-C2N、3Mo-C2N、3Ru-C2N和3Ti-C2N催化剂表面的吸附能分别为−1.27、−2.97、−1.59和−3.73 eV。负值的吸附能表示CO2能稳定吸附在3TM-C2N催化剂表面。C-O键长从1.20~1.42 Å的变化以及O-C-O键角从108.97~128.80˚的弯曲都说明了CO2得到了有效的活化。
为了深入探究CO2在催化剂表面的吸附反应,我们计算了金属三聚体的d带中心和差分电荷密度图。图4(a)显示金属三聚体的d带中心与吸附能呈线性关系,相关系数R = −0.75,与d带中心理论模型符合较好。为了更深入的了解金属三聚体上CO2的吸附作用,我们从三维轨道中提取了alpha(α)态和beta(β)态的d带中心位置如图4(b),图4(c)所示。α态和β态的d带中心变化趋势和总d带中心变化趋势一致,但α态d带中心与吸附能线性关系的相关系数为R = −0.99,相关性更好,说明CO2吸附在催化剂表面时与金属三聚体的α轨道相互作用更强。图4(d)~图4(g)中的差分电荷密度显示,正电荷在金属原子上积累(图中青色部分)。电子由金属原子向CO2分子转移使CO2得到了活化。

Table 1. The adsorption energies ( E ads ), change value of Gibbs free energy adsorbed by CO2 ( Δ G ), bond lengths of C and O atoms in CO2 ( d C − O ) and O-C-O angles of the most stable CO2 adsorption configurations on 3TM-C2N
表1. 3TM-C2N上吸附CO2最稳定构型的吸附能(
)、CO2吸附吉布斯自由能变化值(
)、CO2中C和O原子的键长(
)和O-C-O角度
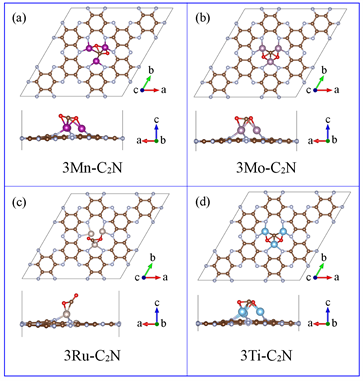
Figure 3. (a)~(d) The stable structure of CO2 adsorbed by the optimized 3TM-C2N
图3. (a)~(d) 优化后3TM-C2N吸附CO2的稳定结构图

Figure 4. (a) The linear relationship between the total d-band center of the transition metal trimer in 3TM-C2N and the adsorption energy of CO2; (b) The linear relationship between the d-band center of the alpha-state orbital of the transition metal trimer and the adsorption energy of CO2 in 3TM-C2N; (c) The linear relationship between the d-band center of the beta-state orbital of the transition metal trimer and the adsorption energy of CO2 in 3TM-C2N; (d)~(g) Charge density differences of CO2 adsorbed by by 3TM-C2N
图4. (a) 3TM-C2N中过渡金属三聚体的总d带中心与CO2吸附能的线性关系图;(b) 3TM-C2N中过渡金属三聚体 态轨道的d带中心与CO2吸附能的线性关系图;(c) 3TM-C2N中过渡金属三聚体β态轨道的d带中心与CO2吸附能的线性关系图;(d)~(g) 3TM-C2N吸附CO2时的差分电荷密度图
3.3. 析氢反应与二氧化碳还原反应的选择性
众所周知,在动力学上容易发生的HER作为副反应阻碍了CO2RR的进行。所以一个好的催化剂能抑制HER是必要的。表1和表2显示,在3Mn-C2N、3Mo-C2N、3Ru-C2N和3Ti-C2N上的氢吸附吉布斯自由能分别为−0.99、−2.44、−0.43和−0.89 eV。
> 0.4 eV说明HER在3TM-C2N催化剂上较难发生 [33] 。同时,
说明*CO2比*H更容易吸附在3TM-C2N催化剂表面从而抑制HER。另外,在第一步质子化过程中,CO2加氢会生成*COOH (*CO2 + H+ + e− → *COOH)和*OCHO (*CO2 + H+ + e− → *OCHO)两个中间体。对于HER来说,也有*H (* + H+ + e− → *H)产生。如图5所示,通过比较
与
的大小(吉布斯自由能较低的选择性较高),3TM-C2N催化剂都落在右下半区域(CO2RR选择性),倾向于生成*COOH或*OCHO。综合以上两点,可以得出3TM-C2N催化剂对CO2RR具有选择性,能够很好地抑制HER的发生。

Figure 5. Free energy changes of the first hydrogenation steps in CO2RR and HER on 3TM-C2N
图5. 在3TM-C2N上发生二氧化碳还原反应和析氢反应第一步加氢反应的自由能变化
Table 2. The Gibbs free energy change ( Δ G ) and adsorption energies ( E ads ) of the most stable H2 adsorption configurations on 3TM-C2N
表2. 3TM-C2N上吸附H原子最稳定构型的吉布斯自由能变化值(
)和吸附能(
)
3.4. CO2还原为CH4
图6显示了在3TM-C2N催化剂上,CO2通过8电子路径生成CH4的中间体吸附结构和自由能分布图。CO2在3Mn-C2N和3Mo-C2N上生成CH4的路径为:CO2 → *CO2 → *COOH → *CO → *COH → *HCOH → *CH2OH → *CH2 → *CH3 → CH4。对于3Mn-C2N,速率限制步骤是*COH → *HCOH,吉布斯自由能变化最大值为−0.44 eV。对于3Mo-C2N,速率限制步骤是*CH3 → CH4,吉布斯自由能变化最大值为−0.97 eV。CO2在3Ru-C2N上生成CH4的路径为:CO2 → *CO2 → *COOH → *HCOOH → *HCO → *HCOH → *CH → *CH2 → *CH3 → CH4,速率限制步骤是*CH2 → *CH3,吉布斯自由能变化最大值为−0.73 eV。CO2在3Ti-C2N上生成CH4的路径为:CO2 → *CO2 → *COOH → *CO → *COH → *HCOH → *CH → *CH2 → *CH3 → CH4,速率限制步骤是*CO → *COH,吉布斯自由能变化最大值为−1.63 eV。

Figure 6. (a) The corresponding stability structure diagram of the reaction intermediate along the reaction path of b; (b) Gibbs free energy distribution diagram of CO2 reduction pathway toward CH4 on 3TM-C2N (numbers 0~8 represent electron transfer numbers)
图6. (a) 反应中间体沿b反应路径的对应稳定结构图;(b) 在3TM-C2N上CO2还原产生CH4的吉布斯自由能分布图(数字0~8代表电子转移数)
3.5. 生成CH4的极限电势
图7给出了CO2在3Mn-C2N、3Mo-C2N、3Ru-C2N和3Ti-C2N上还原为CH4的极限电势(UL)。这里近零的UL表示容易生成该产物。生成CH4的UL按递增排列都为3Mn-C2N < 3Ru-C2N < 3Mo-C2N < 3Ti-C2N。综合来看,3Mo-C2N和3Ti-C2N整体性能较差,
> 0.9 V说明反应需要吸收大量的能量而较难发生。性能最好的催化剂是3Mn-C2N。在3Mn-C2N上生成CH4的极限电势是−0.44 V,表现出较好的催化活性,是四种催化剂中所需能量最低的。分析表明,3TM-C2N催化CO2RR是可以实现的。我们的工作探索了3TM-C2N在较低电极电位的电催化下对CO2深度加氢具有优异的性能。尤其是3Mn-C2N在四种催化剂中性能最好。这一结果表明,C2N负载的过渡金属三聚体催化剂具有良好的CO2RR潜力。

Figure 7. The calculated values of UL (in V) for the production of CO2 to CH4 on 3TM-C2N
图7. CO2在3TM-C2N上还原生成CH4时的UL (V)计算值
4. 结论
总之,本工作通过密度泛函理论计算,设计了3TM-C2N (TM = Mn, Mo, Ru, Ti)的模型作为催化剂,讨论3TM-C2N电催化还原CO2为CH4的催化性能。计算表明,锚定在C2N上的锰、钼、钌和钛三聚体构成了稳定的催化剂。6.67~6.79 eV/atom的内聚能表现出强的稳定性。过渡金属三聚体和C2N之间明显的电荷密度积累、明显的轨道重叠和大量的电荷转移是3TM-C2N整体结构高稳定性的原因。CO2吸附表明,3TM-C2N通过三角形结构和TM活性位点的协同作用,为CO2吸附和初始活化提供了有利的环境。CO2在3TM-C2N上的吸附能在–0.63~–3.10 eV范围内。O-C-O键角的弯曲以及C-O键长从1.20~1.42 Å都说明了CO2的有效活化。吉布斯自由能的计算表明,3TM-C2N对CO2较强的吸附以及对中间体*COOH/*OCHO较低的吉布斯自由能表现出对HER良好的抑制性。极限电势表明,3Mn-C2N表现出最好的催化活性,其将CO2还原为CH4的速率限制步骤是*COH → *HCOH,UL为−0.44 V。
基金项目
本研究得到国家自然科学基金资助(批准号:12174035)。
NOTES
*通讯作者。