1. 引言
DNA酶学性质涉及底物DNA和酶切产物核苷酸,但是二者均难以检测,所以DNA酶学性质研究的关键是酶切产物分离和检测。放射性标记DNA最早作为底物研究DNA酶活性,经DEAE-sephadex吸附、NaCl-NaOAc/urea缓冲液梯度洗脱,通过检测酶切产物放射性探讨外切酶性质 [1] - [6]。但是,放射性标记DNA制备复杂而且危害健康 [5] [6] [7]。此后,荧光染料PicoGreen、SYBR Green I、BEBO用于DNA酶学性质研究 [8] [9],定量检测模板DNA拷贝数 [8] [10] [11] [12]。但是,荧光染料不能定量结合DNA,不是专一性染色酶分子底物,所以特异性低。基于荧光淬灭–激发原理,分子标记用于DNA多态性 [13] [14]、定量PCR [10] [11]、T4DNA连接酶和topoisomerase酶学性质 [15] [16]。特别是Sanger终止法测序使用荧光标记ddNTPs [17] [18],荧光素Cy5-ATP标记double-stranded breaks连接中间物用于激光共聚焦显微镜检测 [19],为荧光DNA扩增、用于酶学性质提供了新途径。
与荧光素Cy5-ATP类似,荧光素Cy5-dATP在激发光649 nm、发射光675 nm时显示红色荧光。dNTPs中加入Cy5-dATP可以扩增荧光Cy5DNA。根据目的DNA分子大小、DNA链中dA碱基个数,在dNTPs中加入一定比例Cy5-dATP可以在荧光DNA中加入定量荧光分子。这种荧光Cy5DNA扩增方便,而且特异性修饰酶分子底物,在酶学性质研究更加优越,但是酶切产物分离–检测是关键。
酶切产物分离可以利用DNA纯化柱解决。其纯化原理是:DNA溶液中加入5倍(×)体积P3缓冲液调节溶液pH值和离子强度 [20],从而将DNA吸附在硅胶材料表面,通过离心得到滤出液(含有P3及酶切产物)。而后纯化柱中央加入30 μL TE洗脱缓冲液,改变硅胶材料pH值和离子强度,从而将吸附DNA重新溶解在TE缓冲液中,通过离心得到洗脱液(含有DNA)。这种原理可以分离酶切产物和底物。
本研究基于上述设想,以加入1/1000 Cy5-dATP的dNTPs扩增5851 bp荧光Cy5DNA pET28a-xyn (xyn为黑曲酶GH11木聚酶基因) [21],用于T5 DNA外切酶(T5exo)酶切反应。首先探讨纯化柱截留DNA所需P3缓冲液的最小用量,进而分离Cy5DNA溶液、Cy5DNA-T5exo反应液,分别得到洗脱液(含有Cy5DNA)和滤出液(含有酶切产物Cy5-dATP),通过酶标仪、激光共聚焦显微镜定量、定性检测,为荧光Cy5DNA扩增、用于DNA酶学性质研究奠定基础。
2. 材料与方法
2.1. 材料
Cy5-dATP (母液浓度1 mM)来自于Thermo Fisher Scientific,高保真Q5 DNA聚合酶,dNTPs,T5 DNA外切酶(T5exo)来自于NEB,正向引物JFV2810 (CAGCCATATGATGAGTGCCG)、反向引物JRV2812 (CATATGGCTGCCGCGCGGCACC,下划线表示正、反向引物间有10 bp同源)、DNA回收试剂盒来自于上海生物工程公司。5851 bp质粒pET28a-xyn (黑曲酶GH11木聚酶基因)由本实验室构建 [21]。
2.2. 荧光Cy5DNA的扩增
5851 bp线性LDNA pET28a-xyn扩增:取1 μL 10 ng/μL pET28a-xyn模板质粒,25 uM正向引物JFV2810、2.5 uM JRV2812,1U Q5 DNA聚合酶,1 μL 10 mM dNTP Mix (终浓度200 μM),10 μL Q5 DNA聚合酶buffer,以水补足50 μL。以双退火程序扩增LDNA [22]:98℃变性2 min,98℃变性20 s,74℃退火15 s,61℃退火15 s,72℃延伸3 min 45 s,72℃延伸10 min,28个循环,4℃保温。扩增60管共3000 μL PCR产物,利用大量–高纯–高浓度DNA的制备方法 [23],在1.55 g/mL氯化铯0.2 mg/mL EB溶液中经76000 rpm密度梯度离心6 h纯化并回收LDNA,用Nanodrop 1000检测浓度(Thermo scientific)。
荧光Cy5DNApET28a-xyn扩增:取1000 ng LDNA pET28a-xyn (2.53×10-4 nmol)为模板,以25 uM JFV2810扩增F链荧光Cy5DNA,1U Q5 DNA聚合酶,1 μL 10 mM dNTP Mix (终浓度200 μM),1 μL 10 μM Cy5-dATP (母液稀释100倍达到10 μM),10 μL Q5 DNA聚合酶buffer,以水补足50 μL反应体系。因为PCR体系中有1 μL 10 mM的dATP,则Cy5-dATP:dATP比值为1:1000。PCR程序为:98℃变性2 min,98℃变性20 s,74℃退火20 s,72℃延伸3 min 45 s,72℃延伸10 min,18个循环,4℃保温。扩增30管共1500 μL PCR产物,利用大量–高纯–高浓度DNA的制备方法 [23],纯化并回收Cy5DNA,用Nanodrop 1000检测浓度。
2.3. 纯化柱截留DNA所需P3缓冲液最小用量
检测纯化柱截留DNA所需P3缓冲液最小用量。按照纯化柱操作说明 [20],含120 ng LDNA的溶液,调整体积为100 μL,分别加入溶液体积1~5倍(×)的P3缓冲液,混匀后加入纯化柱中央,8000 g离心30 s分离得到滤出液(含有核苷酸),而后在柱中央加入500 μL洗涤缓冲液,9000 g离心30 s,并重复一次,而后将纯化柱放入新EP管中,柱中央加入30 μL TE缓冲液,9000 g离心1 min洗脱得到25 μL DNA洗脱液(含有DNA)。用Nanodrop检测滤出液和洗脱液DNA含量和纯度指标,而后电泳检测洗脱液和滤出液DNA条带。
2.4. 荧光Cy5DNA酶切反应纯化柱分离
Cy5DNA-T5exo酶切反应:10 μL切割反应体系中加入360 ng荧光Cy5DNA (9.3 × 10−5 nmol),加入1 μL T5exo (10 U),1×T5exo buffer,37℃酶切反应30 min。阴性对照与酶切反应相同,只是加入120 ng荧光Cy5DNA (3.1 × 10−5 nmol),保温完成后加入1 μL (10 U)热失活T5exo (10% SDS条件下98℃热变性10 min),而后相同条件下定量检测反应液荧光值。
根据纯化柱截留DNA所需最小用量2×P3,将Cy5DNA-T5exo反应液调整为100 μL体积,以2×P3 (200 μL)柱分离,并重复一次,加水补足600 μL滤出液,则每200 μL滤出液对应120 ng Cy5DNA酶切产物。柱中央加入30 μL TE洗脱液,得到25 μL洗脱液加水补足200 μL。
含120 ng 荧光Cy5DNA的溶液调整体积为100 μL,以2×P3柱分离得到600 μL滤出液、25 μL洗脱液加水补足200 μL。酶切产物经过柱分离后得到600 μL滤出液,每200 μL滤出液相当于120 ng荧光Cy5DNA。
为探讨P3对荧光值影响,直接以P3进行2×P3柱分离得到600 μL滤出液、25 μL洗脱液,加水补足200 μL。
2.5. 酶标仪检测Cy5荧光
分别将样品通过2×P3柱分离得到的600 μL滤出液、200 μL洗脱液加入200 μL标准黑色酶标板,以水作为空白对照。以SpectraMax® i3x酶标仪定量检测样品荧光值(Molecular Devices, Thermo Fisher Scientific),因为Cy5dATP为红色荧光,动态方式检测时激发光和发射光波长相差必须大于24 nm,所以选择649 nm激发光、675 nm发射光检测,每30 s采集一次数据,共采集21次荧光值平均。
用IBM SPSS Statistics软件分析各样品荧光值数据显著性,选用软件中“分析”→“比较均值”→“单因素ANOVA”,“两两比较”,选择“LSD (L)、Tukey s-b (K)和Waller-Duncan”参数(Duncan’s multiple range test, P < 0.05),将各样品分为显著性差异的不同组。
检测Cy5DNA与荧光值定量关系时,分别取含有60、120、500 ng Cy5DNA的溶液在200 μL体系中检测荧光值,将荧光值与Cy5DNA浓度(nM)拟合得到线性回归方程,可以通过荧光值计算底物Cy5DNA浓度(nM)。
检测Cy5-dATP与荧光值定量关系时,分别取0.1、0.2、0.4、0.5、0.7、0.9 (0.1~0.9) nM Cy5-dATP检测荧光值,将荧光值与Cy5-dATP浓度(nM)拟合得到线性回归方程,可以通过荧光值计算酶切产物Cy5-dATP浓度(nM)。
2.6. 激光共聚焦显微镜检测Cy5荧光
定性检测样品荧光时,先以70%乙醇溶液超声处理10 min载玻片和盖玻片,镊子取出玻片置于擦镜纸晾干。从酶标仪检测后的滤出液和洗脱液中,分别取出5 μL样品滴加到载玻片上,镊子夹住盖玻片倾斜覆盖液体,避免气泡产生。以A1R HD25激光共聚焦显微镜(Nikon Corporation, Japan),选择638 nm处检测Cy5-dATP样品荧光。
3. 结果与分析
3.1. 荧光Cy5DNA的扩增
荧光Cy5DNA扩增如图1 (上)所示:先以pET21a-xyn质粒为模板、dNTPs为底物、JFV2810和JRV2812为引物反向PCR扩增全长5851 bp线性LDNA。而后以LDNA为模板、JFV2810引物、加入1/1000 Cy5-dATP的dNTPs为底物扩增荧光Cy5DNA。而后,Cy5DNA经T5exo酶切,经DNA纯化柱分离得到酶切产物,进而经酶标仪、激光共聚焦显微镜检测。
对于PCR扩增产物,经过大量–高纯–高浓度DNA制备,分离纯化后共得到74214 ng浓度111.1 ng/μL的LDNA,分离纯化得到39030 ng浓度为130.1 ng/μL荧光Cy5DNA,电泳检测得到5851 bp的LDNA (图1下1、2)和Cy5DNA条带(图1下3、4)。

Figure 1. Amplification and electrophoresis of Cy5DNA. (up) LDNA and Cy5DNA were amplified, digested by T5 DNA Exonuclease, separated by DNA-clean column, and assayed by con-focal microscope and microplate reader. (down) Electrophoresis of Cy5DNA and LDNA recovered by DNA-clean column. M: DNA Marker VI, 1~2: linear DNA, 3~4: fluorescent Cy5DNA, 5~9: filtrate from column with 1~5×P3 buffer recovery of LDNA, 10~14: elution from column with 1~5×P3 buffer recovery of LDNA
图1. Cy5DNA扩增及电泳(上) Cy5DNA扩增模式:依次扩增线性LDNA、荧光Cy5DNA,经T5exo酶切,纯化柱分离、检测(下)纯化柱截留DNA电泳,M:DNA marker VI,1~2:线性LDNA,3~4:荧光Cy5DNA,5~9:1~5×P3缓冲液柱回收LDNA的滤出液,10~14:1~5×P3缓冲液柱回收LDNA的洗脱液
5851 bp LDNA正链有1374个dA (23.5%)、1388个dT (23.7%)、1583个dC、1506个dG碱基,在扩增荧光Cy5DNA时加入1/1000 Cy5-dATP的dNTPs,则每个荧光Cy5DNA分子中引入1.374分子Cy5-dATP,这种荧光分子是随机插入这1374个位置的,引物位置除外。
3.2. 纯化柱截留DNA所需最少用量的P3缓冲液
基于DNA纯化柱回收DNA原理,理论上DNA-T5exo酶切产物随P3缓冲液进入滤出液,未酶切DNA截留在洗脱液。与纯化柱以5×P3回收DNA目的不同 [19],分离酶切产物时要减少P3的稀释作用,所以要探讨截留DNA所需P3最少用量。取LDNA溶液调整为100 μL体积,分别加入1、2、3、4、5倍体积的P3 (1~5×P3),按照纯化柱说明书回收LDNA [20],分别得到滤出液和洗脱液,Nanodrop检测DNA浓度,分析1~5×P3用量截留LDNA效果,从而得到截留DNA P3最小用量。
Nanodrop检测DNA浓度表明,纯化柱以2×P3回收LDNA时,洗脱液DNA浓度为6.8 ng/μL,A260/A230指标为1.94,4×P3洗脱液A260/A230指标2.44,均接近于核酸纯度指标2 (表1)。2×P3滤出液有微量DNA,但其A260/A230纯度指标只有0.28,A260只有0.03,接近3~4×P3滤出液DNA浓度(1.77~1.99 ng/μL),3×P3滤出液A260为负值,5×P3滤出液DNA浓度为负值,说明滤出液DNA是碳水化合物污染的误差。所以,2×P3是纯化柱截留LDNA、稀释酶切产物最小用量。
电泳检测显示,2×P3滤出液没有DNA条带(图1,6),而1×P3滤出液有微量DNA条带(图1,5),而且1~5×P3洗脱液DNA含量接近(图1,10~14)。与其它泳道不同,M和10泳道边缘出现拖尾现象,可能因为5~9泳道滤出液中带有清洗缓冲液,其中异丙醇有展开剂作用。电泳检测与Nanodrop结果相同,显示2×P3纯化柱是截留DNA、分离酶切产物的最少用量。

Table 1. Column recovery of LDNA by 1~5×P3
表1. 1~5×P3柱回收线性DNA
3.3. P3缓冲液不影响荧光值
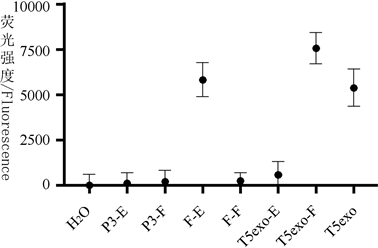
图2. 定量检测荧光值。H2O:空白对照水,P3-E:2×P3经柱回收的洗脱液,P3-F:2×P3经柱回收的滤出液,F-E:Cy5DNA溶液经柱回收的洗脱液,F-F:Cy5DNA溶液经柱回收的滤出液,T5exo-F:T5exo-Cy5DNA经2×P3柱回收滤出液,T5exo-E:T5exo-Cy5DNA经2×P3柱回收洗脱液,T5exo:T5exo-Cy5DNA反应液,a (H2O、P3-E、P3-F、F-F、T5exo-E),b (F-E、T5exo),c (T5exo-F):组间显著性差异(Duncan’s multiple range test, P < 0.05)
纯化柱需要2×P3缓冲液截留DNA、分离酶切产物,i.e.,有2×P3通过纯化柱滤出到酶切产物中,所以需要探讨P3对酶切产物荧光值的影响。以纯化柱单独分离P3分别得到洗脱液和滤出液,酶标仪定量检测显示P3洗脱液、P3滤出液、水空白对照的荧光值相同(图2,a组P3-E、P3-F、H2O),P3洗脱液和P3滤出液荧光值为0,表明P3不影响酶切产物Cy5-dATP荧光值,该结果与P3不含荧光物一致,可以用于定量检测Cy5-dATP。与无荧光相同的a组还包括T5exo-Cy5DNA经2×P3柱回收洗脱液、Cy5DNA溶液经柱回收的滤出液。有显著荧光值的b组包括Cy5DNA溶液经柱回收的洗脱液、T5exo-Cy5DNA反应液,明显高于b组荧光值的c组包括T5exo-Cy5DNA经2×P3柱回收滤出液。
3.4. Cy5DNA荧光定性及定量检测
激光共聚焦显微镜定性检测Cy5荧光素显示,Cy5DNA溶液有荧光(图3,左),而未加入Cy5-dATP的dNTPs扩增LDNA无荧光(图3,中)、Cy5DNA-T5exo反应物也有荧光(图3,右),无法与Cy5DNA荧光区分开来。

Figure 3. Con-focal microscope 20×image of Cy5DNA (left), LDNA (middle), and Cy5DNA-T5exo digestion product (right)
图3. 激光共聚焦显微镜检测Cy5DNA (左)和LDNA (中) Cy5DNA-T5exo酶切产物(右)
Cy5DNA-T5exo酶切反应液检测有荧光(图3右),定量检测荧光值为5387 ± 866 (图2,b组T5exo),接近Cy5DNA洗脱液荧光值(5824 ± 937),阴性对照反应液荧光值为3451 ± 782。Cy5DNA溶液和阴性对照中均有荧光值说明酶切产物无法与底物分离开来,无法确定T5exo是否切割、以及Cy5DNA的切割程度如何。必须经纯化柱以2×P3分离酶切产物,从而与底物Cy5DNA区分开来。
3.5. Cy5DNA-T5exo酶切产物的柱分离、定量检测
Cy5DNA-T5exo反应液经纯化柱以2×P3分离得到600 μL滤出液(酶切产物Cy5-dATP)、200 μL洗脱液。定量检测发现,200 μL滤出液(相当于120 ng Cy5DNA酶切产物)荧光值7573±1527(图2,c组T5exo-F),其洗脱液荧光值0,与水空白对照相同,结果与T5exo酶切Cy5DNA产生游离核苷酸一致 [3] [24]。因为检测荧光值的各种设备所用检测单位不同,只能用绝对单位a.u或数据表示 [25]。
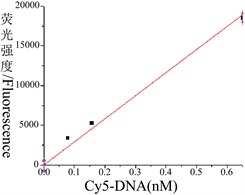
Figure 4. Fluorescence value relationship with Cy5DNA (left), Cy5DNA recovered after DNA-clean column (middle), and fluorescence value relationship with Cy5-dATP (right)
图4. 荧光值与Cy5-dATP定量关系(左) Cy5DNA柱回收洗脱液荧光(中)及荧光值与Cy5DNA定量关系(右)
分别取0.1~0.9 nM Cy5-dATP检测荧光值,Cy5-dATP与荧光值定量关系方程:y = 368957 × x (y为荧光值,x为Cy5-dATP nM浓度,R2为0.996) (图4,左)。根据Cy5-dATP与荧光值定量关系方程,滤出液荧光值7573 ± 1527相当于Cy5DNA浓度为7573 ÷ 368957 ÷ 1.374 × (5851 ÷ 1374) × 2 = 0.127 nM,i.e.,120 ng Cy5DNA经T5exo酶切产物Cy5-dATP荧光值。结果说明T5exo完全切割120 ng荧光Cy5DNA产生了Cy5-dATP核苷酸,并通过纯化柱以2×P3分离到了滤出液中。
含120 ng Cy5DNA的溶液经纯化柱以2×P3分离得到600 μL滤出液、加水补足200 μL洗脱液。定性检测发现Cy5DNA洗脱液有荧光基团(图4,中)。定量检测洗脱液荧光值5824 ± 937,滤出液荧光值0,该结果与纯化柱截留DNA原理一致。
分别取含60、120、500 ng Cy5DNA的溶液检测荧光值,Cy5DNA与荧光值定量关系为:y = 29079 × x (y,x分别为荧光值和Cy5DNA nM浓度,R2为0.992) (图4,右)。根据Cy5DNA荧光值定量关系方程(图3,右),5824 ± 937洗脱液荧光值相当于5824 ÷ 29079 ÷ 1.374 = 0.146 nM Cy5DNA,接近于120 ng Cy5DNA理论计算浓度0.155 nM。因为Cy5DNA长度为5851 bp,正链中有1374个dA碱基,每1000 dATP中加入1.374个荧光Cy5-dATP (dNTPs中加入1/1000的Cy5-dATP用于扩增Cy5DNA,i.e.,每个荧光Cy5DNA中有1.374个荧光Cy5-dATP)。

Figure 5. Con-focal microscope 20×image of T5exo filtration (left), Cy5DNA-T5exo elution (middle) and Cy5DNA filtration (right)
图5. 激光共聚焦检测Cy5DNA-T5exo切割反应滤出液(左),洗脱液(中),Cy5DNA滤出液(右)
定量检测必须与定性检测结果保持一致,定性检测显示Cy5DNA-T5exo滤出液有明显荧光(图5,左),而Cy5DNA洗脱液有明显荧光(图4中)。所以说2×P3纯化柱分离了酶切产物Cy5-dATP与底物Cy5DNA。检测发现Cy5DNA-T5exo洗脱液没有明显荧光,但是有痕量荧光基团(图5中),这种荧光在误差范围内,Cy5DNA滤出液没有明显荧光,但是也有痕量荧光基团(图5右),这种荧光在误差范围内,定量检测荧光值的差异显著性分析中与水、P3滤出液、P3洗脱液没有显著性差异(图2,a组)。
4. 讨论
放射性标记DNA制备过程复杂而且危害健康 [1] - [6],荧光染料PicoGreen、SYBR Green I、BEBO是非特异性荧光染料 [8] [10] [11] [12],不能定量确定一个DNA分子结合多少个荧光分子。这些染料对所有DNA染色,不是专一性染色酶分子底物,所以特异性不高。本研究dNTPs中加入1/1000 Cy5-dATP扩增5851 bp荧光Cy5DNA pET28a-xyn。Cy5DNA优势在于扩增方便、扩增量大、特异性高、灵敏度高,而且可以在每个DNA分子定量加入荧光分子,所以特异性高。酶学性质研究的关键是产物和底物分离,根据纯化柱吸附DNA原理,探讨了纯化柱截留DNA最小用量为2×P3,为荧光值定性和定量检测奠定基础。
dNTPs中Cy5-dATP比例可以控制Cy5DNA荧光强度,进而控制酶切产物荧光强度,提高酶切产物检测灵敏度。5851 bp线性DNA pET21a-xyn正链含有1374个dA碱基,dNTPs加入1/1000 Cy5-dATP,则每个Cy5DNA带有1.374分子Cy5-dATP。120 ng (3.1 × 10−5 nmol) 荧光Cy5DNA荧光值5824 ± 937,其T5exo酶切产物滤出液荧光值提高1.3倍达到7573 ± 1527,与正链1374个dA碱基中加入1/1000 Cy5-dATP一致。3.1 × 10−5 nmol Cy5DNA有等nmol (3.1 × 10−5 × 10−9 × 6.02 × 1023 = 18.7 × 109)个Cy5-dATP,相当于1/1374 (13.6 × 106)个全链标记荧光Cy5DNA分子。表明DNA双螺旋影响Cy5荧光的释放、检测,而酶切产物游离核苷酸有利于荧光释放、检测。这个结果与荧光Cy5DNA浓度、Cy5dATP浓度与荧光值关系一致。如果扩增1000 bp DNA,则需要dNTPs加入1:100的Cy5-dATP使每个DNA带1个荧光分子。与之相比,EB染色检测DNA的最低限度为10 ng,电泳酶切产物呈现大小不一的弥散条带,所以只能检测底物降解量。长度大小不一的DNA结合EB量也不相同,无法定量比较不同大小的DNA。超螺旋DNA与线性DNA结合EB量也不相同。另外,EB与PicoGreen、SYBR Green I、BEBO荧光染料一样不能定量结合DNA。Nanodrop、分光光度计可以检测DNA浓度A260值,但是酶切反应是产物和底物混合物,无法区分产物和底物,同时无法区分dsDNA和ssDNA。
荧光值定量酶切产物时用到标准曲线,结果显示0.1 nM~0.9 nM Cy5-dATP标准曲线合适,定量检测时发现大于0.4 nM Cy5-dATP有光漂白现象,小于0.4 nM则几乎没有漂白现象,i.e.随检测时间增加荧光强度衰减现象,这与荧光分子数量有关,所以检测荧光时需要合适浓度。荧光定量检测时与溶液体积有关,选择酶标板最大200 μL体积能最大限度减少误差。定量检测显示Cy5DNA-T5exo洗脱液和Cy5DNA滤出液与水空白、非荧光DNA对照相同,定性检测确认不含DNA。但是二者有痕量荧光基团(图5,中右),说明纯化柱不能100%截留Cy5DNA,也不能100%滤出Cy5-dATP,显著性分析显示痕量残留在误差范围内(图2,a组T5exo-E,F-F)。纯化柱分离和检测荧光时,样品间平行操作可以去除这种误差。
研究发现与DNA纯化柱不同,蛋白浓缩的超滤管(3 kDa和100 kDa)不能分离Cy5DNA-T5exo酶切产物,可能蛋白浓缩时不考虑小分子物质是否滤出或吸附膜上。另外,纯化柱吸附200 bp以下DNA效率较低 [20],如果酶切产物小于200 bp,需要通过聚丙烯酰氨凝胶检测,垂直电泳提供聚丙烯酰氨的无氧环境。荧光Cy5DNA在酶学性质研究中特异性更高,但是每个样品都需要纯化柱分离荧光Cy5DNA,探讨更方便的分离方法也是荧光Cy5DNA研究的发展方向。
5. 结论
本研究以1/1000 Cy5dATP加入dNTPs扩增5851 bp的荧光Cy5DNA pET28a-xyn,其T5exo酶切产物通过纯化柱以2×P3有效分离,从而与底物Cy5DNA荧光分离开来。在荧光基团Cy5相应激发光649 nm和发射光675 nm波长下,通过激光共聚焦显微镜、酶标仪定性、定量检测酶切产物荧光值,进而分析了Cy5DNA浓度、dATP浓度与荧光值的定量关系,为荧光DNA定量用于DNA酶学性质研究奠定了基础。
基金项目
国家自然科学基金:基于同源/非同源结构置换的木聚糖酶–葡聚糖酶结构元件功能解析(31771915)。
NOTES
*通讯作者。