1. 引言
原发性肝癌是目前全球最常见的恶性肿瘤之一,根据最新研究,在世界范围内发病率排名第六,死亡率排名第三,对人的生命和健康构成了重大威胁 [1] 。原发性肝癌包括肝细胞癌(HCC)、肝内胆管癌及其他罕见类型,而HCC占所有病例85%~90%,我们在本研究中主要关注HCC [2] 。随着医学技术的进步,肝癌现有的治疗方法日新月异。目前肝癌的治疗方法主要包括:手术治疗、肝移植、消融治疗、介入治疗、放射治疗、化学疗法、靶向疗法、免疫疗法。尽管近年来在治疗HCC的方案选择上取得了巨大进步,但HCC起病隐匿,往往发现不及时,错过了最佳治疗时间。因此迫切需要寻找一个新的敏感生物标志物,一方面,它能有效帮助医生制定正确治疗方案;另一方面,它有助于鼓励患者加强对异常指标的监测,及时发现异常,并尽早治疗 [3] 。
多项研究表明,表观遗传学失调在肿瘤发生发展中起着重要的作用 [4] 。DNA甲基化是研究最多的,是在DNA甲基化转移酶(DNA methyltransferase, DNMT)的作用下,由SAM提供的甲基团与胞嘧啶5号碳原子可逆的结合形成5-甲基胞嘧啶。相比于基因突变,表观调控具有可逆性,这使得DNA甲基化在恶性肿瘤中可作为潜在的治疗靶点 [5] 。以往有研究表明,异常的DNA甲基化与HCC的发生、诊断、治疗和预后有着密切联系 [6] [7] 。尽管DNA甲基化与HCC的预后研究较多,但是将基因表达与甲基化联合分析的却较少报道。高甲基化状态的抑癌基因及低甲基化状态的原癌基因,都被认为与肿瘤的发生和发展相关 [8] [9] 。本研究结合基因表达与DNA甲基化数据,筛选出甲基化驱动基因,即低甲基化状态的原癌基因表达量升高,探寻出肝癌肿瘤标志物的基因。
后基因组技术的出现以及全球公开数据集的显著发展,为研究肝癌的发病机制提供了工具,使得研究在单个阵列中同时表达数千个基因的mRNA成为可能,利用生物信息学方法可以识别与肿瘤相关的潜在生物标志物和信号通路 [10] 。许多研究使用全基因组数据集来识别肝癌的诊断和预后分子标记,特别是来自癌症基因组图谱(TCGA, The Cancer Genome Atlas)研究网络的HCC数据集 [11] 。本研究从TCGA数据库中下载肝细胞肝癌基因表达数据和甲基化数据,并将甲基化谱与基因表达谱进行匹配。筛选出癌与癌旁组织中表达水平有差异的基因和甲基化基因,进一步筛选出低甲基化状态下高表达基因ZC2HC1A。该分子在肝癌中作用尚不明确,为研究该分子与肝癌的关系,本研究在mRNA水平上利用多个公共数据库资料验证其表达差异、预后评估、临床应用价值以及免疫浸润之间的潜在价值。进一步探讨ZC2HC1A是否可以作为HCC的肿瘤标志物。
2. 资料和方法
2.1. 数据下载
我们从癌症基因组图谱(TCGA) (https://portal.gdc.cancer.gov/repository)中选择工作流程类型HTSeq-FPKM,下载HCC中ZC2HC1A的基因表达谱和相对应的临床数据,该数据库包含374个肿瘤组织样本和50个正常组织样本,包括年龄、性别、生存时间、生存状态、分级、分期、肿瘤大小(T)、淋巴结(N)、转移(M)。排除样本中不全数据,如生存信息不足以及临床病理特征信息不明确的。
2.2. ZC2HC1A的表达与甲基化的相关性
DNMIVD (http://119.3.41.228/dnmivd/index/)数据库可分析基因启动子区的甲基化水平与相应基因表达之间的相关性 [12] 。基于此数据库分析ZC2HC1A基因启动子区甲基化程度在肝癌组织和正常组织中的表达差异情况,以及分析ZC2HC1A启动子区甲基化程度与其表达量的相关性(Spearman)。
2.3. 癌组织和癌旁组织ZC2HC1A基因差异性表达
首先,使用Limma包提取单基因表达数据,并使用中位值将数据分为高表达组和低表达组(基因表达大于/小于中位值的样品被认为是高/低表达组)。第二,计算威尔科克斯检验的p值,其中p < 0.05表明肝癌组织和癌旁组织的基因表达有显著差异。Beeswarm软件包用于绘制散点图进行差异表达分析。最后,为了进一步证明结论,使用Perl脚本获取癌旁组织和胃癌组织的配对样本,并绘制配对样本的差异表达分析图。
2.4. 基因表达与临床特征的相关性
从TCGA数据集收集了大量的临床病理信息,包括性别、年龄、肿瘤淋巴结转移(TNM)分期、分级和病理分期。逻辑回归用于测试基因表达和临床特征之间的相关性,其中p < 0.05被认为具有统计学意义。
2.5. 生存分析
在生存分析中,所有癌旁组织样本被移除,并且仅保留肝癌组织样本。基于高表达组和低表达组,Kaplan-Meier用于绘制所选基因的存活分析曲线,其中p-value值0.05用作统计阈值。
2.6. 单因素和多因素COX回归分析
单变量和多变量的Cox比例风险模型用于计算95%置信区间和风险比。单变量Cox分析模型用于比较临床特征和生存率之间的关系。多变量Cox分析模型用于评估ZC2HC1A的表达和临床因素(性别、年龄、分级、分期和TNM分期)如何影响OS。同时绘制森林箱线图。将ZC2HC1A表达的p值< 0.05设定为阈值。
2.7. 富集分析
我们使用从TCGA获得的RNA序列数据进行了GSEA分析。基因集富集分析(GSEA)是一种计算方法,它可以确定一个先验定义的基因集是否在两种生物状态之间显示出统计上显著的、一致的差异 [13] 。排列的数量被设置为1000。利用GSEA,我们分析了KEGG途径,以研究ZC2HC1A可能的生物学功能。要被认为具有统计学意义,富集结果必须满足两个条件;FDR < 0.050,p值 < 0.050。
2.8. 免疫细胞浸润分析
肿瘤免疫细胞浸润评估(Tumor Immune Estimation Resource, TIMER)是一个综合数据库(https://cistrome.shinyapps.io/timer/),用于预测基因的表达量与各类型免疫细胞浸润情况的关系 [14] 。我们用TIMER评估了ZC2HC1A的表达和六种肿瘤浸润免疫细胞(TIICs)之间的潜在关系。此外,为了评估样本集之间基因表达的相对变化,我们使用了一种基于基因表达的反卷积算法,称为CIBERSORT ((http://cibersort.stanford.edu/) [25] 。使用CIBERSORT,我们测量了22个TIICs的免疫反应,以评估它们与HCCZC2HC1A表达的相关性,并揭示TIICs之间的相关性。我们还分析了ZC2HC1A对免疫系统微环境的影响,我们使用了374个肿瘤样本,并将其分为两组。以上结果均满足p值 < 0.05。
2.9. 综合分析
GEPIA是一个web服务器和一个在线数据库(http://gepia.cancerpku.cn/),分析了来自GTEx和TCGA的RNA-Seq表达数据,其中包括8,587个正常样本和9,736个肿瘤样本 [15] 。我们从TCGA获得了样本,并使用GEPIA分析了HCC总生存率与ZC2HC1A表达之间的关系。此外,使用疾病状态作为变量的箱线图用于计算ZC2HC1A的差异表达。ZC2HC1A被输入数据库以获得K-M存活图。p值 < 0.05被认为具有统计学意义。
3. 结果
3.1. ZC2HC1A基因表达量与甲基化水平的相关性
我们使用DNMIVD数据库检索获得ZC2HC1A启动子区的甲基化数据。与正常组织相比,肝癌组织中ZC2HC1A启动子的甲基化程度与相应正常组织具有显著差异(p = 8.33e−06) (图1(a)),而且ZC2HC1A在肝癌中的表达与甲基化修饰程度显著负相关性(r = −0.41, p = 1.71e−18) (图1(b)),提示ZC2HC1A的在肝癌中表达可能受到的ZC2HC1A的甲基化调控。
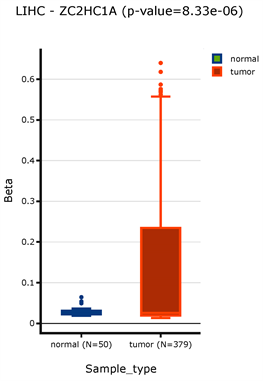 (a)
(a)  (b)
(b)
Figure 1. (a) The difference of the methylation degree of ZC2HC1A in liver cancer tissues and paracancer tissues; (b) The correlation between the expression of ZC2HC1A in liver cancer and the degree of methylation
图1. (a) ZC2HC1A的甲基化程度在肝癌组织和癌旁组织中的差异性;(b) ZC2HC1A在肝癌中的表达与甲基化程度的相关性
3.2. ZC2HC1A的表达在肝癌组织和癌旁的差异性
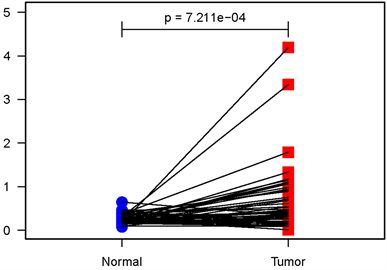
根据分析得出癌组织中ZC2HC1A表达水平的中值明显高于癌旁组织,因此认为癌组织中ZC2HC1A高表达(图2(a))。这个结论也可以从成对组织样本的差异表达分析中得出(图2(b)),其中线连接同一患者的癌旁组织和肝癌组织。表明ZC2HC1A的表达水平在肝癌组织中高度表达。
 (a)
(a) 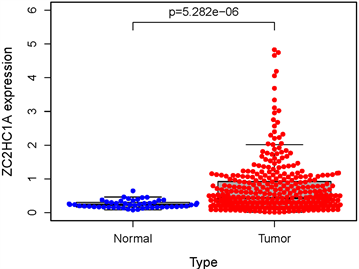 (b)
(b)
Figure 2. Expression analysis of ZC2HC1A. (a) The scatter plot of differential expression analysis of ZC2HC1A in two types of tissues; (b) The differential expression analysis map of paired samples. Blue dots represent the paracancerous tissues, and red dots represent the GC tissues
图2. ZC2HC1A的表达。(a) ZC2HC1A在两种组织中差异表达分析的散点图;(b) 配对样本的差异表达分析图。蓝点为癌旁组织,红点为胃癌组织
3.3. ZC2HC1A表达和临床特征的相关性
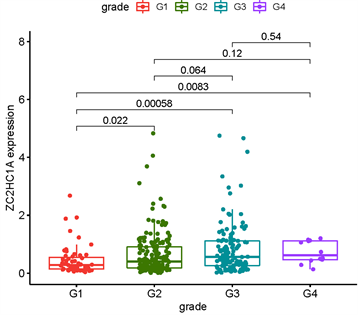
根据来自TCGA的临床病理信息,我们评估了HCC患者的ZC2HC1A表达水平与各种临床病理参数之间的关系。ZC2HC1A表达增加与肿瘤组织学分期(I vs III的p值 < 0.05)、分级(I vs II,III,IV的p值 < 0.05)和T (I vs III的p值 < 0.05)显著相关。这些结果表明,与ZC2HC1A表达水平较低的患者相比,ZC2HC1A表达水平较高的HCC患者更容易患分级和分期更晚的肿瘤(图3)。
 (a)
(a) 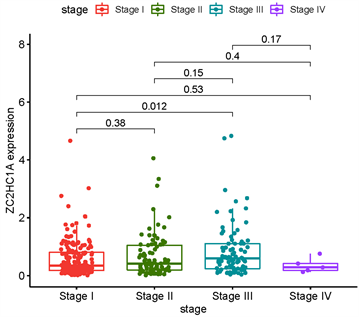 (b)
(b)  (c)
(c)
Figure 3. Expression of ZC2HC1A correlated significantly with histological grade (a) and clinical stage (b) and T stage (c)
图3. ZC2HC1A的表达与临床特征的相关性,分级(a),临床分期(b)和T分期(c)
3.4. ZC2HC1A生存分析
在ZC2HC1A表达联合甲基化的生存分析结果(图4(a))中,红线代表高甲基化低表达组,黑线代表低甲基化高表达组。从图中的结果可以看出,低甲基化高表达组的存活率明显低于高甲基化低表达组。在ZC2HC1A表达的生存分析结果(图4(b))中,红线代表高表达组,黑线代表低表达组。从图中的结果可以看出,高表达组的存活率明显低于低表达组。综合分析得出,ZC2HC1A低甲基化高表达时预后差。根据ROC曲线(图4(c))分析,ZC2HC1A的表达具有较好的预后能力,因为ROC曲线显示ZC2HC1A表达预测1年、3年、5年生存率的AUC分别为0.741、0.613、0.595。
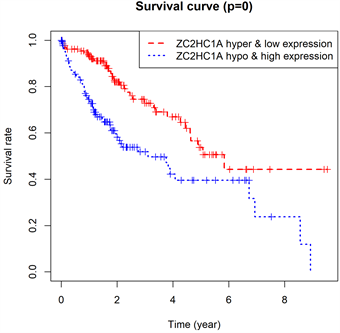 (a)
(a) 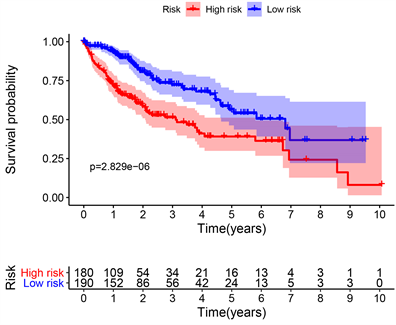 (b)
(b) 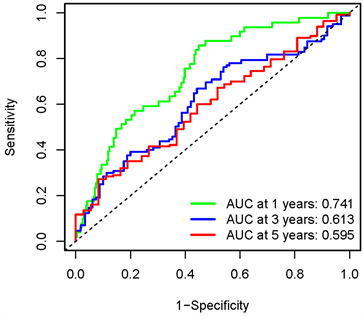 (c)
(c)
Figure 4. (a) The overall survival rate of the hypermethylation low expression and hypomethylation high expression groups of ZC2HC1A; (b) The overall survival rate of the high and low expression groups of ZC2HC1A; (c) The time-dependent ROC analysis for risk score in the TCGA cohort
图4. (a) ZC2HC1A高甲基化低表达组和低甲基化高表达组的总生存率;(b) ZC2HC1A高表达组和低表达组的总生存率;(c) TCGA队列中风险评分的时间依赖ROC分析
3.5. 单因素和单因素COX回归分析
单变量生存分析中,部分因素包括stage分期(HR = 1.865, p < 0.001)、T (HR = 1.804, p < 0.001)、M (HR =3.850, p= 0.023)、ZC2HC1A表达(HR =1.753, p < 0.001) 与OS显著相关。多变量生存分析中,ZC2HC1A表达(HR =1.758, p < 0.001)与OS显著相关。由森林箱线图(图5)描述的单变量和多变量Cox分析也表明ZC2HC1A表达与OS显著相关(p值 < 0.001)。因此,ZC2HC1A可作为HCC的独立预后因素。
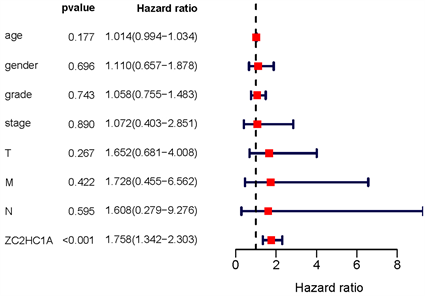
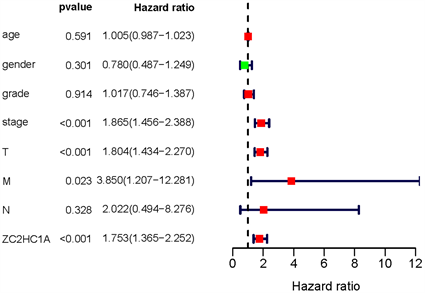
Figure 5. Forest boxplot for Univariate and multivariate Cox analysis of the correlation between ZC2HC1A expression and OS as well as other clinical features
图5. ZC2HC1A表达与OS及其他临床特征相关性的单因素和多因素Cox分析森林箱线图
3.6. 富集分析
通过基因集富集分析来探索ZC2HC1A的潜在生物学功能,并用于分析ZC2HC1A基因的高表达组和低表达组。图6显示了基因组百科全书(KEGG)途径分析的结果,前五个途径与ZC2HC1A表达正相关,前五个途径与ZC2HC1A表达负相关。这些结果表明ZC2HC1A的表达与TGF-β信号通路,细胞周期,WNT信号通路,癌症通路,VEGF信号通路显著相关。
3.7. ZC2HC1A表达与肿瘤浸润性免疫细胞的关系
我们使用TIMER分析了ZC2HC1A表达和免疫浸润细胞水平之间的相关性。如图7(a)所示,ZC2HC1A的表达与B细胞(p-value = 2.34 × 10−7)、CD8+T细胞((p-value = 3.27 × 10−10)、CD4+T细胞((p-value = 1.61 × 10−15)、巨噬细胞((p-value = 1.99 × 10−27)、树突状细胞((p-value = 3.17 × 10−19)和中性粒细胞(p-value = 1.09 × 10−22)的水平呈正相关(如图7(a))。同时,我们还发现了22种免疫细胞在不同的HCC患者中的占比不同(如图7(b)),体现了HCC的异质性。此外,我们将肿瘤样本根据ZC2HC1A表达分为高表达组和低表达组,使用CIBERSORT (Profiles of immune cell infiltration in head and neck squamous carcinoma)确定了22种免疫细胞在两组的水平,从图7(c)中我们发现,幼稚B细胞、CD8+T细胞、滤泡辅助T细胞、活化NK细胞、单核细胞、M0巨噬细胞、静息DC细胞和静息肥大细胞在ZC2HC1A高表达组和低表达组中具有明显差异性(p < 0.05)。与低表达组相比,高表达组M0巨噬细胞增多(p值 < 0.001),而活化的自然杀伤细胞、CD8+T细胞、静息的肥大细胞、幼稚B细胞减少(p值< 0.05)。上述结果表明,ZC2HC1A可能通过影响免疫细胞浸润调控肝癌的发生和发展。
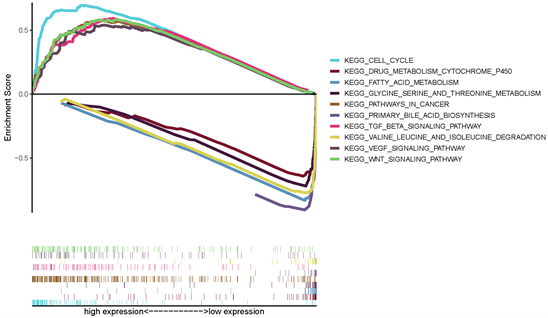
Figure 6. KEGG pathway shows five positive and five negative correlated groups
图6. KEGG通路显示5个正相关组和5个负相关组
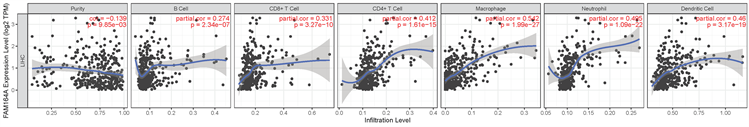 (a)
(a) 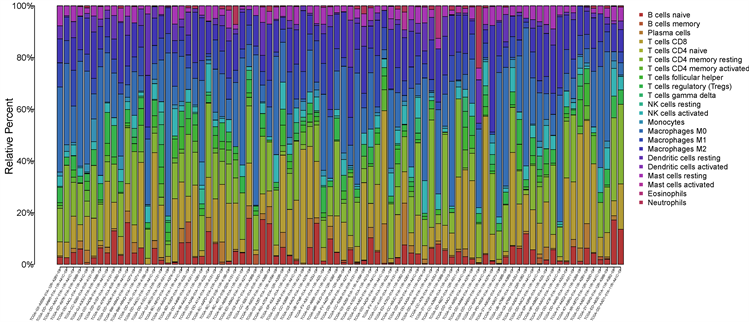 (b)
(b) 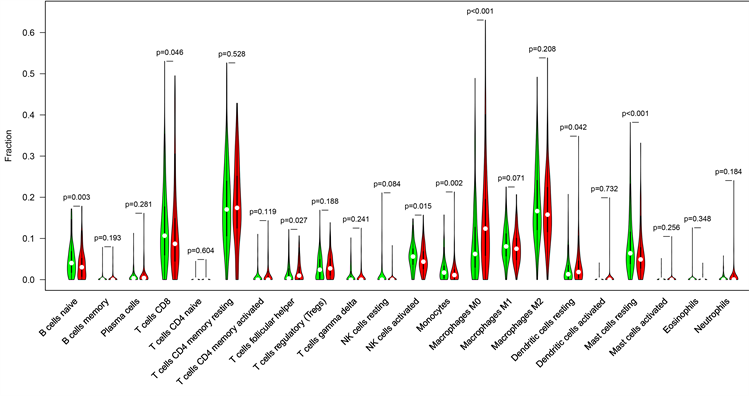 (c)
(c)
Figure 7. (a) Correlations between ZC2HC1A expression and immune infiltration levels; (b) The varied proportions of 22 subtypes of immune cells in different tumor samples; (c) The varied proportions of 22 subtypes of immune cells in high and low ZC2HC1A expression groups in tumor samples
图7. (a) ZC2HC1A表达与免疫浸润水平的相关性;(b) 22种亚型免疫细胞在不同肿瘤样本中的比例不同;(c) 肿瘤标本中ZC2HC1A高、低表达组22种亚型免疫细胞比例的变化
3.8. 数据验证
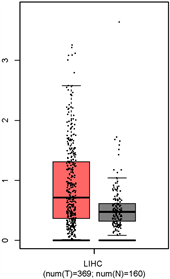
使用GEPIA数据库,我们发现当HCC组与正常组相比时,HCC的ZC2HC1A基因表达显著增加(p值 < 0.01,|Log2FC| > 1) (图8(a)),并且ZC2HC1A基因水平与不良的OS存在强相关性(p值 < 0.001,图8(b))。
 (a)
(a) 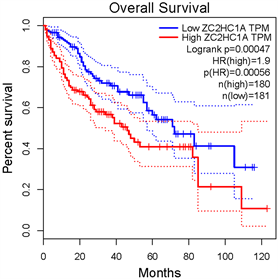 (b)
(b)
Figure 8. (a) ZC2HC1A mRNA expression levels in normal and HCC tissues, as obtained from GEPIA; (b) Levels of ZC2HC1A mRNA expression and overall survival based on data obtained from GEPIA
图8. (a) 从GEPIA中获得ZC2HC1A mRNA在正常和HCC组织中的表达水平;(b) 基于GEPIA数据的ZC2HC1A mRNA表达水平和总生存率
4. 讨论
虽然肝细胞癌治疗的诊疗技术上已经取得了显著的进步,但肝细胞癌的预后整体仍不太理想 [16] 。近年来,原发性肝癌流行率继续增加 [17] [18] 。所以发现肝细胞癌生物标志物对于临床治疗来说意义重大,它不仅可以预测罹患肝癌的风险以及肿瘤进展情况,更可以用来在患者没有明显临床症状的阶段确诊疾病,并为患者治疗提供有效帮助 [19] 。许多先前的研究提供了肝癌生物标志物的信息 [20] [21] [22] ,但到目前为止,能够预测肝癌患者生存的生物标志物仍然是无效的。但是迄今为止,还没有报道ZC2HC1A对HCC的作用,因此,需要更好的了解ZC2HC1A在HCC的作用及其潜在的预后价值,以及它作用的分子机制。
在本研究中,我们对TCGA数据库进行挖掘,将肝癌的表达数据、甲基化数据以及临床数据整理并分析,找到了甲基化驱动基因ZC2HC1A,发现ZC2HC1A的表达和甲基化均存在明显的差异性,且两者呈现明显的负相关。随后,又进一步分析了ZC2HC1A表达和HCC患者的临床特征、生存预后的相关性。我们的研究表明,当ZC2HC1A高表达时,与较差的组织学分级、晚期临床阶段、较差的TNM分期、较短的存活时间有密切的关系,这说明在ZC2HC1A高的患者中,HCC更易复发,侵袭和转移能力变强,并预示着预后不良。此外,单变量和多变量Cox回归分析提示,在排除其他临床病理因素影响的情况下,ZC2HC1A仍然是可以独立预测不良OS的因素。总的来说,这些发现为使用ZC2HC1A表达作为HCC的诊断和预后生物标志物提供了强有力的支持。
在HCC的发生发展过程中,信号转导通路的激活或抑制在肝癌细胞的生长调控中起着至关重要的作用,信号通路的改变会导致细胞增殖和凋亡的失控,发生癌变。通过研究信号通路中不同因子以及蛋白质的表达对HCC的调控,有助于我们找到广泛而有效地HCC生物靶点和新的靶向治疗药物。由于关于ZC2HC1A的文献很少,我们基于GSEA进一步研究了ZC2HC1A的功能,结合GSEA的表现,我们发现了wnt,VEGF,TGF,cancer,cell-cycle信号通路在ZC2HC1A高表达表型显著富集,这可能是ZC2HC1A高表达影响肝癌不良预后的几种潜在机制。
在HCC中,高度血管化是典型特征之一,而VEGF是一种强的血管内皮因子,与VEGFR结合后激活并触发VEGF信号通路,使之上调,刺激内皮细胞增殖,诱导肿瘤周围产生新生血管 [23] ,同时促进肿瘤的生长及转移。目前,已发现很多的靶向治疗药物可以作用于VEGF/VEGFR信号通路,如贝伐珠单抗(Bevacizumab),索拉菲尼,Brivanib、Sunitinib、Cediranib等也作为VEGFR抑制剂应用于临床中 [24] 。TGF-β信号通路在HCC的发生、发展中具有双重作用,早期抑制肿瘤细胞生长,晚期则促进肿瘤细胞侵袭转移 [25] 。TGF-β是肝癌细胞最重要的上皮细胞间质化(EMT)激活剂 [26] [27] ,在TGF-β诱导肝癌EMT过程中,TGF-β还可使肝癌细胞获得肿瘤干细胞的特性,而且,TGF-β还能刺激肝癌肿瘤微环境中巨噬细胞向M2型转化,并分泌高水平的促炎因子,促进肝癌侵袭转移 [28] 。Wnt蛋白是一种伴脂质修饰的分泌型糖蛋白,其主要通过Wnt/平面细胞极性、Wnt/Ca2+和Wnt/β-Catenin信号通路3种途径传导信号,而后者被视为经典Wnt信号通路,主要通过激活其下游靶基因而影响HCC的生成与发展 [29] 。在有Wnt信号时,与受体结合而活化胞内DVL,抑制了糖原合成酶激酶-3β (GSK-3β)活性,阻止了β-catenin的降解,β-catenin聚集在细胞浆内并转位至细胞核,与核转录因子TCF/LEF结合后使靶基因如C-myc、cyclin D1、Survivin等激活并表达,导致细胞的异常增殖,促进HCC进程 [30] [31] [32] 。肝癌通过多种机制和信号通路最终导致肿瘤生成,而仍有许多未知因素有待探究,弄清各个信号通路之间的影响机制以及找到针对信号通路合适的靶点,对HCC的治疗及研究方向的展开起着至关重要的作用。可以从多个通路间的共性及作用模式角度出发,有助于高效、广谱HCC靶向药物研究的深入开展。
在既往的研究中,对肿瘤微环境(TME)进行了很多的研究,而且在肝癌的发生发展中起到了重要作用。TME是个复杂的系统,包括T细胞、B细胞、树突细胞、巨噬细胞和NK细胞的亚群等一系列的免疫细胞 [33] 。在我们的研究中,分析了ZC2HC1A对肿瘤免疫微环境中的免疫细胞的影响。首先,我们通过TIMER数据库分析了ZC2HC1A与免疫浸润的关系,结果显示与巨噬细胞、中性粒细胞、树突状细胞和T细胞具有明显相关性。同时,还分析了22种肿瘤免疫浸润细胞(TIICS)与HCC的关系。TIICS在同种癌症不同患者中存在明显的异质性,通过CIBERSORT分析,我们发现ZC2HC1A表达水平与巨噬细胞M0、休眠的DC细胞、滤泡辅助T细胞呈现正相关,与B细胞、CD8+T细胞、活性NK细胞、单核细胞、肥大细胞呈现负相关。
TME中的肿瘤相关免疫细胞包括抗肿瘤免疫细胞和促肿瘤免疫细胞两大类 [34] 。研究表明,未分化的巨噬细胞(M0)很容易被细胞因子和其他刺激诱导分化为M1和M2表型,三者可以同时存在于肿瘤中 [35] [36] [37] ,并且促进肿瘤的发生发展。在我们的研究中,我们仅发现ZC2HC1A高表达时M0比例增高,且与M1和M2巨噬细胞相比,M0巨噬细胞与患者肿瘤进展程度和预后、生存率明显呈现为负相关。这些升高的M0细胞能一直促肿瘤,进而降低免疫治疗的效果。在肿瘤免疫细胞中,CD8+T细胞是负责杀伤肿瘤细胞,是肿瘤免疫应答最主要的效应细胞 [38] ,而且CD8+细胞含量与患者的预后呈正相关 [39] 。肿瘤免疫细胞NK是一种固有免疫淋巴细胞,通过抗原非特异性的方式识别癌细胞,并通过释放细胞毒性因子或FasL/TRAIL途径诱导其凋亡,发挥抗癌功能。在包括HCC的多种肿瘤中,NK计数或其细胞毒活性的增加显示了更好的预后 [40] 。表明NK细胞也是抗肿瘤免疫细胞 [41] 。肥大细胞根据肿瘤类型,进展情况在抗肿瘤免疫中发挥双重作用 [42] 。在我们的研究中发现,肥大细胞具有抗肿瘤特性。通过对肿瘤患者免疫功能状态的评估,对动态观察肿瘤的生长转移及患者预后有一定参考价值。一般而言,免疫功能正常者预后较好,晚期肿瘤或有广泛转移的患者其免疫功能常明显低下。综上所述,ZC2HC1A能通过影响免疫细胞浸润调控肝癌的发生发展,当ZC2HC1A高表达时,抗肿瘤细胞CD8+T细胞、NK细胞、肥大细胞消耗过多,而促肿瘤M0巨噬细胞明显升高,然而ZC2HC1A在免疫调节中的具体作用以及是否可以作为免疫治疗的生物标志物或联合治疗的靶点有待进一步研究。
通过我们的研究和发现表明,ZC2HC1A可能是一个致癌基因,在HCC中当发生去甲基化后,出现表达量升高,对肝癌患者预后明显产生的不良影响,并且在免疫浸润细胞的调节和募集中起着关键作用,因此推测ZC22HC1A可能是一个关键的免疫检查点。近年来兴起的新型肿瘤过继免疫细胞治疗,给肿瘤免疫治疗领域带来新的亮点 [43] 。根据我们的研究,是否可以使用过继性免疫细胞治疗将有待进一步的研究。
总之,这是一项肝细胞癌新生物标志物的研究,ZC2HC1A可作为诊断和治疗HCC病的有效工具,并可能有助于使生物标记疗法成为未来肝病治疗的有希望的选择。虽然我们目前的研究对肝癌中ZC2HC1A的作用机制已经有了初步的了解,但有一些局限性。第一,本研究是一项回顾性研究,应进行前瞻性研究弥补回顾性研究的局限性。第二,公共数据库TCGA中缺乏临床因素,如患者药物和/或手术治疗的具体细节,也会影响患者的预后。第三,所有的TCGA数据都是从西方国家收集的,在地域和人种上有一定的差异。第四,解释免疫浸润的方法和适当的临界值需要标准化。最后,我们的结果是基于公共数据库和计算机算法完成,因此,有必要使用基础实验、动物模型及多中心临床实验来进一步验证我们的发现。虽然这项研究有一些局限性,但它确实为研究肝癌ZC2HC1A的功能提供了线索,并为HCC的治疗提供了靶点和潜在的预后标记。
NOTES
*通讯作者。