1. 引言
基因调控网络(Gene Regulatory Network, GRN)是由细胞内基因之间相互作用构成的网络。节点代表基因,节点间的连边代表基因之间的调控关系。它是一个抽象的概念,但可以利用数学表达式来建立基因调控的过程,从而帮助人们理解细胞内基因之间的关系。重构基因调控网络属于逆向工程问题,目前该领域的学者提出了很多用于研究基因调控网络的数学模型 [1] 。例如最简单的、离散的布尔网络(Boolean Networks)模型 [2] ;一种概率描述的贝叶斯网络(Bayesian Networks)模型 [3] 以及微分方程(Differential Equation)模型 [4] 等。
基因表达数据反映的是直接或间接测量得到的基因转录产物 mRNA在细胞中的丰度,它包含的基因活动信息能够反映当前细胞的状态,采用反向工程的方法分析基因表达数据可以还原基因的调控网络。基因表达数据可以分为时间序列基因表达数据和静态基因表达数据,它们都是基因表达水平的数据矩阵。时间序列基因表达数据记录了生命活动周期或不同状态下连续变化的基因表达水平,反映了基因调控网络的动态变化,可以通过相应的算法推断基因之间的调控关系 [5] ;静态基因表达数据记录了稳定状态下细胞内的基因表达水平,包含了基因间的调控信息,直接反映了基因之间的调控关系,可以通过基于信息论等方法建立基因调控网络 [6] [7] 。静态基因表达数据具有高维度、小样本的特点,即与大量的基因相比,样本数量较少。对于这一困难多种方法被提出验证。Fujita等对于样本数据量低于基因数量的问题,提出SVAR方法建模基因表达调控网络 [8] ;Barrera等利用互信息降维的方法,通过计算基因之间的互信息,最终选择基因之间互信息较高的基因恢复基因调控网络 [9] ;Székely等扩充了样本数据量,通过距离相关性概念将m个样本数据扩充为m2个样本数据。通过增加样本数据 [10] ,可以提高模型的预测性能。
真实生物体基因间的调控关系错综复杂,模型识别真正的边显得十分重要。而模型在恢复基因调控网络拓扑结构过程中可能会出现真实基因中没有调控关系但被模型错误的预测为有调控关系的情况,即预测结果中出现比较多假阳性边的情况。因此需要降低假阳性边的数量,提高模型预测的准确率。Fujita等提出通过统计检验控制错误发现率的方法 [8] 。罗霄提出通过DSWLasso逐步删除恢复的基因调控网络中的弱调控关系的方法 [11] 。
本文在此基础上进行研究,将基因调控网络的恢复问题转化为识别方程参数的问题。通过假设检验判断基因间是否存在调控关系,此时将基因调控网络拓扑结构恢复问题转化为分类问题进行解决。并采取适当的方法减少结果中出现的假阳性边的数量。
2. 方法
回归分析是用来确定两种或两种以上变量之间相互依赖的定量关系的一种统计方法。本文所研究的基因调控网络包含多个自变量,并且因变量和自变量之间呈线性关系,因此可以利用多元线性回归模型判断基因调控网络中基因间的调控关系。
2.1. 基因调控网络线性回归模型
基于距离样本数据采用回归理论建立基因调控网络线性回归模型,以此判断基因i与其他基因之间是否存在调控关系,模型表示为:
(1)
其中
表示除第i个基因外,其余
个基因的距离样本向量,
代表误差,满足相互独立且分布服从
。
样本数据代入(1)中,则(1)可以改写为:
(2)
恢复基因调控网络拓扑重构解释为从可测变量
和
中估计
,则转化为求解多元线性回归方程(2)的参数
的问题。在这里采用对每个基因进行单独回归分析的方法,用最小化误差的平方和估计每个基因的
,得到预测的邻接矩阵
。损失函数利用均方误差的形式,同时它也是目标函数:
(3)
为确定模型的参数
,同时使预测值
尽可能地接近真实值
,等价于求
的最小值。由于目标函数(3)中包含要求解的参数
,因此在求解(3)最小值的过程中便可以将模型(2)中的参数
求解出来,在这里我们选取最小二乘法求解。用最小二乘法估计
,只需要令其对
的一阶偏导数为0,偏导数为0的点是(3)的最小值点,求得:
(4)
(4)就是
的最小二乘估计,该方法适用于
的逆矩阵存在
时的情况。此时
是
的无偏估计,无偏性保证了最小二乘估计具有较好的结果。
2.2. 基因表达数据处理
基因之间的调控关系隐含在基因表达数据里,需要分析该数据从而揭示基因调控网络的拓扑结构。而现实难以收集到大量的基因表达数据,但对于(4)式,当
时,
不可逆,此时最小二乘估计失效,因此需要扩充样本量。本文根据Székely的方法引入距离相关性来扩充样本量。假设有N个基因,实验收集到这N个基因在m个样本上的表达水平,记为
,其中
。根据距离相关性和
可以生成一个矩阵
,
包含m2个元素,各元素定义为:
其中
表示
范数。对矩阵
中心化可得对称矩阵
,
中元素定义为:
将
中下三角元素重新组合为一列,可得到一个包含
个元素的列向量
,
就是对
原始基因表达数据
的扩充,由原来的m个样本扩充为M个样本,因此达到了扩充样本基因表达数据的目的,称
为距离样本数据。
2.3. 显著性检验
2.3.1. 基因调控网络线性回归模型的可行性
本文采用多元线性回归方程预测基因i和其它基因间的调控关系,建立的恢复模型为(2)。但基因i和其它基因之间是否为线性关系是未知的,因此需要判断线性关系是否成立。通过对模型(2)进行整体检验来判断回归系数是否不全为0,如此便可说明其他基因是否对基因i有显著影响。如果所有回归系数为0,则代表它们对基因i没有线性调控关系,因此这相当于检验原假设
是否成立:
对应的备择假设为:
如果有充分的理由说明
不成立,那么意味着基因i与其它
个基因之间的线性关系成立。对模型整体检验选取
检验。在
为真时构造检验统计量:
其中
表示自由度为
的
分布,取显著性水平为
。我们选定
,当
时拒绝原假设
,认为基因i与其它
个基因之间有线性关系,说明利用基因调控网络线性回归模型恢复拓扑结构是可行的。
2.3.2. 基因调控关系的判定
模型整体检验说明基因i与其它
个基因之间的线性关系成立,但是不能确定某个基因j对基因i是否有调控关系。因此需要对回归系数进行检验,即讨论基因与基因之间是否有调控关系。
本节采用的基因调控矩阵元素为0和1,其中0代表两个基因之间没有调控关系,1表示两个基因之间有调控关系。利用模型(2)判断哪些基因之间存在调控关系,等价于寻找非0的
。因此为检验基因i与其他基因之间是否存在调控关系,建立如下假设:
如果有充分的理由说明
不成立,那么就可以接受
,认为基因i与基因j之间存在调控关系。选取的假设检验方法为t检验。已知
,当假设
为真时构造检验统计量:
其中
表示自由度为
的t分布,
为
的第j个对角线元素,
为
的无偏估计。取显著性水平为
。我们选定
,当
时拒绝原假设
,接受备择假设
,认为
,说明基因j对基因i的作用显著,即确定基因j对基因i有调控关系;反之,当
时不拒绝原假设
,则认为
,说明基因j对基因i的作用不显著,即确定基因j对基因i没有调控关系。
通过对模型(2)基于距离样本数据利用最小二乘估计得到初始的预测矩阵后,接着对回归系数进行假设检验便可以确定
中元素为1的位置,
中元素为1意味着基因之间有调控关系,此时模型只保留了对基因i有显著作用的调控基因。因此可根据构建的模型确定具有调控关系的边,即完成了恢复基因调控网络的拓扑结构。
对模型恢复的基因调控网络拓扑结构进行整体评估,定义评价指标:
为模型未识别率,其中
表示基因间有调控关系而模型没有识别出的个数,
表示基因间真实又调控关系的个数;
为假阳性率,其中
表示基因间没有调控关系而模型识别为有调控关系的个数,
表示基因间没有调控关系的个数;
为误报率,其中
表示模型识别基因间有调控关系的个数。
2.4. 控制假阳性结果的数量
t检验的拒绝域为
,而由于随机性等因素的干扰,在进行判断时可能出现两
种错误,假阴性错误和假阳性错误。若出现原假设成立,但计算得到的
落入拒绝域
中的错误,称为假阳性错误(第Ⅰ类错误)。在Neyman-Pearson假设检验中,可以要求假阳性率在可接受的范围内,设置的检验水平
就是限制犯假阳性错误的概率,通过
达到对单重假设检验的统计推断的错误控制。在单个假设检验中假阳性率是可控的,但是对于整体多个假设而言,假阳性结果会随着检验次数的增加而增大,出现超出预设范围的情况。因此需要对回归系数整体进行多重检验校正,减少出现假阳性结果的次数。选择控制FDR的BH(Benjamini-Hochberg)法,该方法可以将假阳性率控制在统计显著水平
以下。调整
值控制FDR,过程如下:
对于假设检验
,
表示第
个检验的原假设。将所有检验的
值从小到大排序生成顺序数
。令
,定义
其中v代表检验次数。当
时,拒绝前k个假设,认为基因之间有调控关系。
3. 模型应用及恢复结果
3.1. DREAM数据集上的实验
通过以上分析可以根据模型(2)推断基因调控网络,为了验证其可行性,本节在DREAM(Dialogue for Reverse Engineering Assessments and Method)提供的静态基因表达数据上进行实验。采用DREAM3数据集,数据集可在https://gnw.sourceforge.net/dreamchallenge.html下载。DREAM3数据集提供了静态基因表达数据和确定的基因调控网络,本节实验选取数据集InSilicoSize10-Ecoli1-null-mutants,它包含10个基因,将基因标号为
。该数据集对应的网络结构如图1所示。其中红线代表基因之间是抑制作用,蓝线代表基因之间是激活作用。本文不区分基因之间的抑制和激活作用,主要关注基因之间是否存在调控关系。
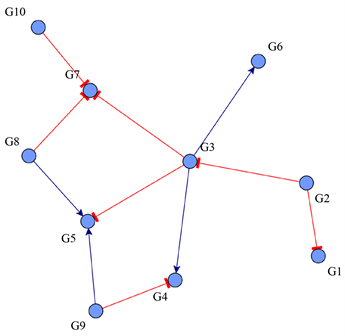
Figure 1. Topology of gene regulatory network
图1. 基因调控网络拓扑结构
由于实验会受到随机性的影响,为了确保模型预测的稳定性,结果为多次实验的平均。每次实验对距离样本采用随机抽样用于模型(2)来构建基因调控网络,每次抽样循环多次并取平均值作为最终结果。
3.2. 实验结果及讨论
3.2.1. 基因调控网络线性回归模型的可行性
DREAM3提供了确定的基因调控网络拓扑图,本文只考虑基因之间是否存在调控关系,因此调控矩阵中的元素只有0,1两种。数据集InSilicoSize10-Ecoli1-null-mutants中仅包含11个基因表达数据,首先对其进行扩充得到包含66个样本数据距离样本,从中随机选取不同长度的样本用于恢复拓扑结构。
对每个基因在不同样本数据时的模型(2)进行检验以判断利用其恢复基因调控网络拓扑结构是否可行,表1包含了在不同数据下对每一个模型进行整体检验的F值。
在
情况下,由表1以及Friedman检验表可知,对于G8,G9,G10基因,由于
,因此不拒绝原假设
,认为回归方程的所有系数为0,与真实的基因调控网络一致;对于另外7个基因,由于
,因此拒绝原假设
,认为每一个基因
与其它基因之间有线性关系,说明利用线性回归模型预测基因调控矩阵是可行的。

Table 1. F-value of the overall model test
表1. 模型整体检验的F值
3.2.2. 基因调控关系的判定及控制假阳性结果的数量
对于原假设
和备择假设
,采用t检验可以判定矩阵
中位置为1的元素,即可以判定基因之间的调控关系。
图2显示曲线呈下降趋势,模型的未识别率E在逐渐降低,说明随着数据量M的增加,模型识别到的连接个数逐渐增加。但初始样本数据会限制模型对基因调控网络拓扑结构的恢复能力,当
时,
,此后距离样本再增加不会明显提高模型的恢复能力。
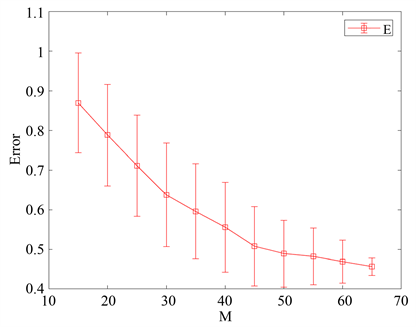
Figure 2. For the gene regulatory network linear regression model (2), the unrecognized rate E with its standard deviation under different sample data size M
图2. 对于基因调控网络线性回归模型(2),不同样本数据量M下的未识别率E及其标准差
由图3可知,
,说明模型(2)在恢复基因间的调控关系时出现较多的假阳性边,即出现较多实际基因没有调控关系但模型错误的判断为有调控关系的结果。图4是在
时对模型(2)应用控制FDR的BH法后恢复的基因调控网络中假阳性边的情况。由图可知
,此时假阳性率低于0.05。与图3相比,模型预测的假阳性由5条减少为1条,达到了控制模型识别假阳性边的个数的目的。因此对于在构造基因调控网络出现假阳性边较多的情况,可以采用控制FDR的BH法来减少出现模型恢复过程中出现假阳性边的次数。
不失一般性,在不同数据量时均可对整体多个假设采用FDR校正。由图5可得,对于不同的样本数据量M,FDR校正后假阳性率较之前会降低,达到了对构建的基因调控网络假阳性边数量控制的目的,假阳性率始终在(0, 0.05)的范围之内。

Figure 3. For the gene regulatory network linear regression model (2), the restored gene regulatory relationship when M = 40. The solid line represents the true gene regulatory relationship, and the dashed line indicates the edges where there is no regulatory relationship between genes but the model identifies as having a regulatory relationship
图3. 对于基因调控网络线性回归模型(2),M = 40时模型恢复的基因调控关系。实线表示真实的基因调控关系,虚线表示基因间不存在调控关系但模型识别为有调控关系的边
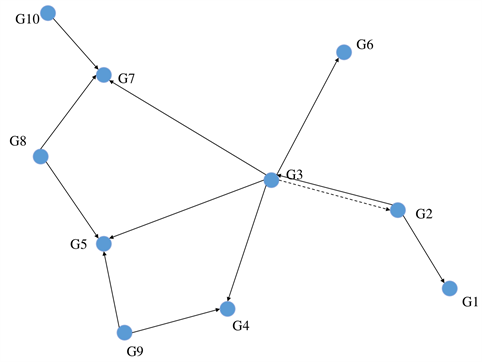
Figure 4. For the gene regulatory network linear regression model (2), the restored gene regulatory relationship when M = 40 after FDR correction. The solid line represents the true gene regulatory relationship, and the dashed line indicates the edges where there is no regulatory relationship between genes but the model identifies as having a regulatory relationship
图4. 对于基因调控网络线性回归模型(2),经过FDR校正后在M = 40时模型恢复的基因调控关系。实线表示真实的基因调控关系,虚线表示基因间不存在调控关系但模型识别为有调控关系的边
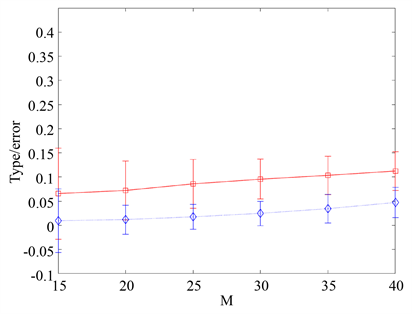
Figure 5. For the gene regulatory network linear regression model (2), the probability and standard deviation of false positive error under different sample data size M. The red line and blue line respectively represent FPR values before and after FDR correction
图5. 对于基因调控网络线性回归模型(2),不同样本数据量M下犯假阳性错误的概率及其标准差。红线蓝线分别代表FDR校正前和校正后的FPR的值
由于假阳性错误和假阴性错误在相同的样本数据量下不能同时增大或减小,只有增加样本数据量才可以使两种错误同时减少,因此采用FDR校正降低模型识别假阳边的数量后,不可避免的会增加假阴性边的个数。但由图6可知随着数据量的增加,FDR校正前后模型出现假阴性错误的差距越来越小。

Figure 6. For the gene regulatory network linear regression model (2), the unrecognized rate E with its standard deviation under different sample data size M. The red and black lines represent the unrecognized rate E before and after correction, respectively
图6. 对于基因调控网络线性回归模型(2),不同样本数据量M下的未识别率E。红色线和黑色线分别代表校正前和校正后的未识别率E
4. 结论
基因调控网络存在于生物体内,是由细胞内基因之间相互作用构成的网络,可以利用数学表达式建立基因调控的过程,这可以帮助我们了解细胞内基因之间的关系。静态基因表达数据记录了稳定状态下细胞内的基因表达水平,可以反映基因之间的调控关系,但是其样本数较少,本文对此介绍了扩充样本数据产生距离样本的方法,并基于距离样本根据回归理论建立了基因调控网络线性回归模型,通过最小二乘估计和假设检验来确定哪些基因之间存在调控关系。接着应用控制FDR的BH法有效地减少了模型识别假阳性结果的数量,提高模型预测的准确率。最后选取DREAM3数据集验证模型的可行性,并在M = 40时直观展现了该方法降低假阳性边的有效性。
基金项目
本文工作由国家自然科学基金(No. 11502062)支持。
NOTES
*通讯作者。