1. 背景
胰腺导管腺癌是胰腺最常见的恶性肿瘤,发病率与死亡率几乎相等 [1] [2] 。据全球癌症统计报道,2022年胰腺癌新发病例在所有肿瘤中排第七位,而死亡病例在所有肿瘤中位列第三 [1] 。预计至2030年左右,胰腺癌将超过胃癌,结直肠癌成为肿瘤导致死亡的第二大主要原因。由于早期症状不明显且缺乏有效的早期诊断指标,大约80%的病人在确诊时已发生了远处转移 [3] 。吉西他滨对延长进展期胰腺癌病人生存期作用有限 [4] 。因此寻找新的治疗方法对改善胰腺癌预后十分重要。
近几年来,以免疫检查点抑制剂为代表的免疫治疗在结直肠癌,胃癌及黑色素瘤治疗取得重大突破 [5] 。然而,由于胰腺癌免疫细胞丰度较低和免疫抑制的肿瘤微环境,免疫治疗在胰腺癌的疗效十分有限 [6] 。由于肿瘤组织内部的抑制性,不同病人对免疫治疗的应答也不相同 [7] 。因此,建立胰腺癌免疫亚型对改善胰腺癌病人预后,精准诊疗及提高免疫治疗疗效十分重要。
本研究通过一致性聚类方法,基于免疫微环境评分的结果,对145例胰腺癌患者重新分组。随后,通过对两组胰腺癌患者的差异分析,功能富集分析,蛋白互作网络分析来寻找调控胰腺肿瘤微环境的关键基因。为区分不同胰腺癌免疫亚型提供了理论支持,同时也为开发胰腺癌免疫治疗新靶点提出了新思路。
2. 材料与方法
2.1. 数据来源
从GEO数据库GSE 71729芯片获取了145例胰腺癌患者(GSM 1844109-GSM 1844245)的转录组测序数据。转录组数据标准化采用每千个碱基的转录每百万映射读取的fragments (FPKM)的方法。
2.2. 免疫亚型
使用“ESMIMATER”R包计算145例胰腺导管腺癌患者的免疫平分,基质评分及肿瘤纯度。基于免疫平分,基质评分及肿瘤纯度,使用“consensusclusterplus”R包对145例胰腺导管腺癌患者进行一致性聚类。使用pearson相关距离的pam聚类,对80%的样本重新抽样10次。使用经验累积分布函数图来确定最佳的聚类数量。
2.3. 差异分析
利用lmFit函数进行多元线性回归,进一步使用eBays函数进行计算t统计量,通过对标准误差的经验贝叶斯节制来计算差异表达的对数,最终获得每个基因的差异显著性。
2.4. 富集分析
将从KEGG rest API 下载了京都基因 与 基 因 组百科全书 (Kyoto Encyclopedia of Genes and Genomes, KEGG)通路富集分析基因注释作为背景,将帅选出差异基因映射至背景合集中,使用clusetrprofiler R包进行富集分析。最小基因设定为5,最大基因集设定为5000。p < 0.05且FDR < 0.25认为有统计学意义。
2.5. 蛋白互作网络
本研究运用cytoscape中的cytohubba插件进行分析,本研究跟根据连接度Degree进行排序,筛选Degree前30的基因作为本研究的hub基因。
2.6. 单细胞测序
我们使用“seurat”R包进行对16例胰腺导管腺癌患者单细胞测序数据进行质控过滤,检测到基因数小于400,超过6000,线粒体比例超过15%的细胞被移除。随后,我们采用Findvirablefeatures函数,选取vst方法,挑选前1500高变基因进行下游分析。运用主成分分析方法对1500个高变基因进行降维。采用tsne对主成分分析结果进一步降维。用“singleR”R包进行注释。使用seuratR包注释细胞类型。
3. 结果
3.1. 免疫亚型鉴定
首先,我们使用“ESTIMATE”R包计算145例胰腺癌患者的免疫评分,肿瘤评分及基质评分。随后,基于这些患者的肿瘤微环境评分,我们使用“consensusclusterplus”R包对这些患者进行一致性聚类。CDF曲线显示,k = 2时分型结果最好(图1)。其中亚型1免疫评分,基质评分较高,命名为热肿瘤,亚型2免疫评分较低,命名为冷肿瘤。
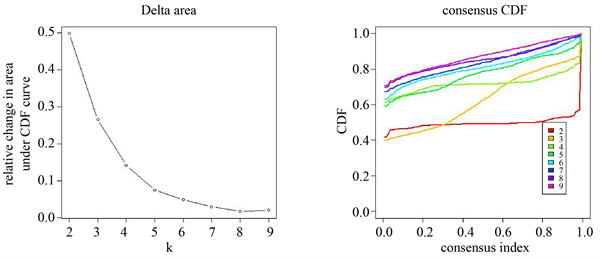
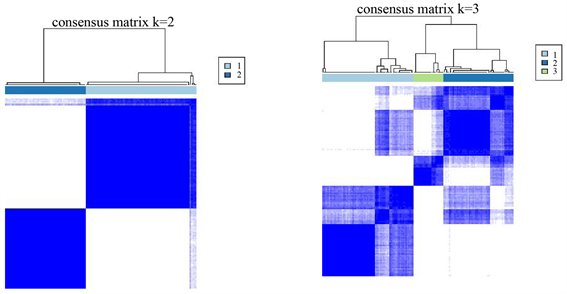
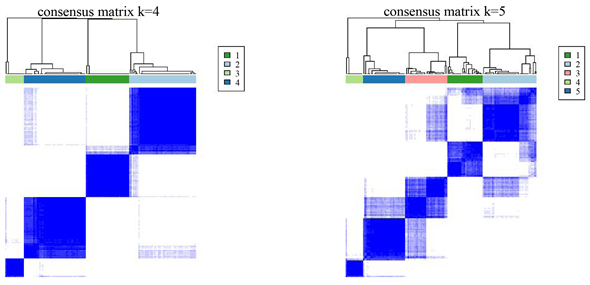
Figure 1. Identification of the PDAC immune subtype
图1. 胰腺导管腺癌免疫亚型鉴定
3.2. 差异分析
随后,我们采取“limma”包对这两种不同的免疫亚型进行差异分析,来寻找两种免疫亚型之间的差异基因,adj p < 0.05 且logFC > 1设置为筛选差异基因的阈值。在这两种免疫亚型之间,我们会总共鉴定到298个差异基因(见图2)。
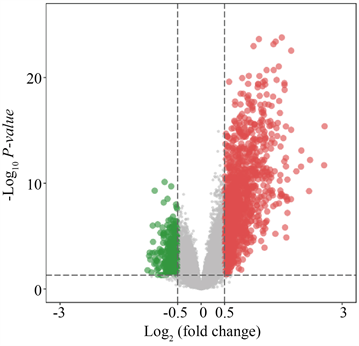
Figure 2. Volcano map for differential expressed analysis of different immune subtypes
图2. 不同免疫亚型差异分析火山图
3.3. 功能富集分析
我们对这298个差异基因进行了KEGG富集分析。富集分析结果表明这些差异基因主要富集在Cell adhesion molecules,Intestinal immune network for IgA production,Hematopoietic cell lineage,Complement and coagulation cascades,Chemokine signaling pathway,Antigen processing and presentation,Cytokine-cytokine receptor interaction,Th1 and Th2 cell differentiation,Leukocyte transendothelial migration,Th17 cell differentiation,ECM-receptor interaction,NF-kappa B signaling pathway,T cell receptor signaling pathway,Natural killer cell mediated cytotoxicity,Human T-cell leukemia virus 1 infection等免疫及纤维化相关通路,说明这些基因可能在调控肿瘤微环境发挥了重要作用,可能成为预测胰腺癌免疫治疗疗效,提高胰腺癌患者对免疫治疗应答率的重要靶点(图3)。
3.4. 蛋白–蛋白互作网络分析
运用cytoscape中的插件cytoHubba进行基因–基因相互作用网络分析,根据节点在网络中的接连接进行排名,确定了前30个基因:C1QA,CSF1R,C1QB,THBS1,LUM,ITK,COL5A1,COL3A1,COL1A1,CD28,CD3G,CD8A,TYROBP,FBN1,COL1A2,DCN,CD247,PIPRC,FN1,LCP2,CFP,BTK,CCR5,MMP9,CXCR4,C1QC,ITGB2,VCAN,CXCL12,CD2。这30个基因的相互作用见图4。
3.5. 单细胞测序检测hub基因表达
为了评估这些基因是如何调控肿瘤免疫微环境,我们从GSE 155698下载了16例胰腺导管腺癌患者的单细胞测序数据。选取前1500个高变基因进行下游分析(图5(a))。在胰腺癌中,总共鉴定出9种细胞:T细胞,B细胞,内皮细胞,肿瘤细胞,纤维细胞,单核细胞,中性粒细胞,NK细胞和组织干细胞(图5(b))。使用“seurat”包中AddModuleScore函数对这30个hub基因在各细胞类型进行打分,结果表明这些细胞在肿瘤细胞中密度最高(图5(c))。
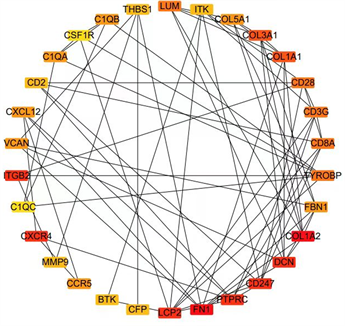
Figure 4. Protein-protein interaction network
图4. 蛋白–蛋白互作网络
 (a)
(a) 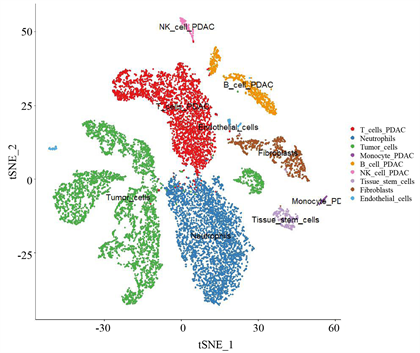 (b)
(b)  (c)
(c)
Figure 5. Single-cell sequencing for detection of hub gene expression. (a): Top 1500 highly variable genes. (b): Classify cell populations using the tSEN algorithm. (c): Hub Gene density map
图5. 单细胞测序检测hub基因表达。(a) 前1500高变基因;(b) 通过tSEN算法对细胞群分类;(c) hub基因密度图
4. 讨论
免疫治疗,特别是免疫检查点抑制剂的出现,开创了肿瘤治疗的新时代。大量研究表明,免疫细胞丰度,特别是CD8T细胞浸润水平与病人预后及免疫治疗疗效密切相关 [8] [9] 。由于肿瘤组织内部的异质性,不同病人对治疗的敏感性不同。本研究的目的是建立一种新的胰腺导管腺癌免疫亚型,为胰腺肿瘤的免疫治疗提供指导。
我们通过“ESTIMATE”R包对145例胰腺癌患者肿瘤微环境内免疫浸润及纤维化程度进行评估。随后使用“consensusclusterplus”R包对这145例胰腺癌患者进行重新分组,建立胰腺癌免疫亚型。对这两种免疫亚型进行差异分析,共鉴定出298个差异基因。KEGG富集分析结果表明这些差异基因主要与Cell adhesion molecules,Intestinal immune network for IgA production,Hematopoietic cell lineage,Complement and coagulation cascades,Chemokine signaling pathway,Antigen processing and presentation,Cytokine-cytokine receptor interaction,Th1 and Th2 cell differentiation,Leukocyte transendothelial migration,Th17 cell differentiation,ECM-receptor interaction,NF-kappa B signaling pathway,T cell receptor signaling pathway,Natural killer cell mediated cytotoxicity,Human T-cell leukemia virus 1 infection等免疫,纤维粘连的通路相关。随后通过蛋白–蛋白互作网络,筛选出排名前30的hub基因,这些基因可能是调控肿瘤微环境的关键基因。
CSF1与外周血单核细胞向肿瘤微环境的迁移有关。与CSF1R结合后,CSF1促进单核细胞向巨噬细胞分化,并向促瘤的M2巨噬细胞表型发展 [10] 。有研究表明,在胰腺癌小鼠模型中,靶向CSF1/CSF1R轴可以重塑肿瘤微环境,提高胰腺癌小鼠对免疫检查点抑制剂的敏感性 [11] 。然而,一项II期临床研究表明,CSF1R阻断并不能改善晚期PDAC患者对吉西他滨的敏感性(NCT 03336216)。CSF1R阻断主要是抑制巨噬细胞的极化,促进细胞毒性T细胞的浸润,增加免疫检查点如PD-L1和CTLA4的表达 [11] 。因此,CSF1抑制剂提高胰腺癌病人对免疫治疗敏感程度仍需进一步评估。同时,有报道称CSF1表达水平可以预测非小细胞肺癌的免疫疗法的疗效 [12] 。CSF1预测胰腺癌免疫治疗疗效的能力仍需评估。
在单细胞测序层面,我们对这30个hub基因表达水平进行了评估。基因集评分结果表明,这些基因在肿瘤细胞表达水平最高。肿瘤微环境在胰腺癌进展中发挥了重要作用,免疫抑制的肿瘤微环境可能是胰腺癌对免疫治疗应答率低的主要原因 [13] 。胰腺癌微环境由基质细胞和免疫细胞组成。有助于胰腺癌进展的基质细胞主要是胰星状细胞(PSCs)、调节性T细胞(Tregs)、骨髓源性抑制细胞(MDSCs)和肿瘤相关巨噬细胞(TAMs)。这些细胞和癌细胞可以分泌细胞外成分,如细胞外基质(ECM)、基质金属蛋白酶(MMP)、生长因子和转化生长因子-β (TGFβ),以维持微环境。胰腺癌微环境有两个主要特点:严重纤维化和广泛的免疫抑制 [14] 。胰腺癌肿瘤微环境是指胰腺癌细胞的生存环境,主要由胰腺星状细胞,肿瘤相关成纤维细胞,免疫细胞与细胞外基质组成。本质上来讲,胰腺导管腺癌处于一个纤维结缔组织广泛沉积的肿瘤微环境内。广泛纤维细胞募集免疫抑制细胞,造成了胰腺肿瘤细胞免疫逃逸。广泛纤维化,缺血乏氧,免疫抑制的肿瘤微环境为肿瘤细胞的生长提供了条件。同时,纤维黏连的基质纤维压迫血管,限制了化疗药物向肿瘤细胞投递,使其对化疗药物产生耐受。这两个特点可以促进胰腺癌细胞增殖,通过直接抑制抗肿瘤免疫力来逃避免疫监视,或诱导免疫抑制性细胞增殖和转移。据此,我们推测这些hub基因主要定位于肿瘤细胞,可能与它们调控胰腺癌免疫抑制与纤维化的肿瘤微环境有关。它们可能是增强胰腺癌免疫治疗疗效的潜在靶点。
本研究不足之处在于所有数据均来自公共数据库,完整的临床数据难以获得,无法评估肿瘤微环境与生存,预后,肿瘤分型及各项生化指标的关系。
总之,我们通过建立胰腺导管腺癌的免疫亚型以及之后的转录组学,单细胞转录组学分析,初步探索了不同胰腺癌免疫亚型的分子调节机制。基于蛋白互作网络和单细胞测序分析,我们鉴定了30个调控肿瘤细胞与免疫,纤维细胞互作的hub基因,可能成为治疗胰腺癌的潜在靶点。
NOTES
*第一作者。
#通讯作者Email: xxmczwz@qdu.edu.cn